一种含有阿托伐醌与伊维菌素的组合物及其途
本发明属于药物,具体涉及一种具有协同增效作用的抗疟组合物。
背景技术:
1、
2、伊维菌素(ivermectin,ivm)是一种广谱、高效、低毒的体内外寄生虫杀灭药物,其主要的作用靶点为无脊椎动物肌肉和神经细胞的谷氨酸门控氯化物(glucl)离子通道(lynagh t,lynch jw.ivermectin binding sites in human and invertebrate cys-loop receptors.trends pharmacol sci.2012;33(8):432-41.),其通过高亲和力结合阻止glucl离子通道闭合,导致氯离子内流及细胞去极化,最终致机体的麻痹或死亡。伊维菌素已被广泛应用于根除盘尾丝虫病和淋巴丝虫病的大规模给药(mass drugadministration,mda)。由于其对按蚊的强致死作用,近年来伊维菌素被考虑用于疟疾传播防控的mda,并在与其它抗疟药联合应用的mda临床试验中显著降低野生按蚊的存活率与人群的疟疾发病率(dabira ed,soumare hm,conteh b,et al.mass drug administrationof ivermectin and dihydroartemisinin-piperaquine against malaria in settingswith high coverage of standard control interventions:a cluster-randomisedcontrolled trial in the gambia.lancet infect dis.2022;22(4):519-528;foy bd,alout h,seaman ja,et al.efficacy and risk of harms of repeat ivermectin massdrug administrations for control of malaria(rimdamal):a cluster-randomisedtrial.lancet.2019;393(10180):1517-1526.)。实验室研究表明伊维菌素对伯氏疟原虫肝内期具有显著抑制作用,在鼠疟模型可达到与伯氨喹相近的红外期疟原虫抑制疗效(mendes am,albuquerque is,machado m,et al.inhibition of plasmodium liverinfection by ivermectin.antimicrob agents chemother.2017;61(2):e02005-16.)。在体外评价模型中,其对食蟹猴疟原虫肝内期裂殖体和休眠体的发育也有抑制作用(vanachayangkul p,im-erbsin r,tungtaeng a,et al.safety,pharmacokinetics,andactivity of high-dose ivermectin and chloroquine against the liver stage ofplasmodium cynomolgi infection in rhesus macaques.antimicrob agentschemother.2020;64(9):e00741-20.)。此外,在体外试验中其对恶性疟原虫红内无性期和有性期的发育同样有显著抑制作用(de carvalho lp,sandri tl,josétenório de meloe,et al.ivermectin impairs the development of sexual and asexual stages ofplasmodium falciparum in vitro.antimicrob agents chemother.2019;63(8):e00085-19.)。伊维菌素对恶性疟原虫的抑制作用被认为与其阻断疟原虫信号识别颗粒组分或某些其它核蛋白的核质穿梭有关(panchal m,rawat k,kumar g,et al.plasmodiumfalciparum signal recognition particle components and anti-parasitic effectof ivermectin in blocking nucleo-cytoplasmic shuttling of srp.cell deathdis.2014;5(1):e994.)。但在随机对照的人体感染疟疾病因性预防临床试验中,单独口服伊维菌素0.4mg/kg体重剂量未能达到有效的预防作用(metzger wg,theurer a,pfleiderer a,et al.ivermectin for causal malaria prophylaxis:a randomisedcontrolled human infection trial.trop med int health.2020;25(3):380-386.)。
3、阿托伐醌(atovaquone,ato)为辅酶q的竞争性抑制剂,可选择性地结合于疟原虫细胞色素b,抑制其线粒体电子传递链并破坏经线粒体膜电位,对红内期和肝内期疟原虫皆有抑制作用,但对肝内期休眠体无活性(nixon gl,moss dm,shone ae,etal.antimalarial pharmacology and therapeutics of atovaquone.j antimicrobchemother.2013;68(5):977-85.)。由于阿托伐醌单药治疗会导致疟原虫快速的产生药物抗性,目前临床主要以与氯胍组成固定比例复方的形式用于无并发症恶性疟的治疗和旅行者的疟疾预防。但其在柬埔寨的使用导致了阿托伐醌抗性恶性疟的快速出现(whoguidelines for malaria,14march 2023.geneva:world health organization;2023)。
4、药物抗性是全球疟疾防控面临的最大挑战。抗药性疟原虫的不断传播与扩散已导致传统一线疟疾治疗药物,如氯喹、氯胍、乙胺嘧啶、磺胺多辛-乙胺嘧啶和甲氟喹,相继失效,无法在大多数的疟疾流行地区获得90%以上的临床疗效。自2001年被世界卫生组织推荐为抗药性疟疾流行国家疟疾治疗的首选一线药物以来,以青蒿素为基础的联合疗法(artemisinin-based combination therapy,act)已成为了当前最有效和临床应用最广泛恶性疟一线治疗药物。然而,随着越来越广泛的临床施用,表现为疟原虫清除速度减慢或act治疗失败的青蒿素抗性已在东南亚大湄公河流域地区出现广泛流行(world malariareport 2022.geneva:world health organization;2022;mathenge pg,low sk,vuongnl,et al.efficacy and resistance of different artemisinin-based combinationtherapies:a systematic review and network meta-analysis.parasitol int.),且近年来已在印度和非洲等其它流行地区出现(das s,saha b,hati ak,et al.evidence ofartemisinin-resistant plasmodium falciparum malaria in eastern india.n engl jmed.2018;379(20):1962-1964;balikagala b,fukuda n,ikeda m,et al.evidence ofartemisinin-resistant malaria in africa.n engl j med.2021;385(13):1163-1171)。恶性疟原虫青蒿素或act抗性的进一步发展与传播将给全球疟疾根除带来严峻挑战。为应对这一挑战,具有不同作用机制的新型抗疟药是当前的迫切需求。基于既往的疟原虫抗药发展的经验,世界卫生组织推荐以固定剂量复方的形式开发抗疟新药,以期防止或延缓疟原虫药物抗性产生、发展。此外,药物对疟原虫生命周期其它阶段的影响,如配子体的形成与存活、药物的半衰期、临床疟原虫血症的清除效率和临床施用剂量,也被视为优先研发复方药物的考虑因素。
5、基于此,提出本发明。
技术实现思路
1、本发明提供了一种含有阿托伐醌或其前药或药学上可接受的盐作为第一活性成分,伊维菌素或其类似物作为第二活性成分的组合物。
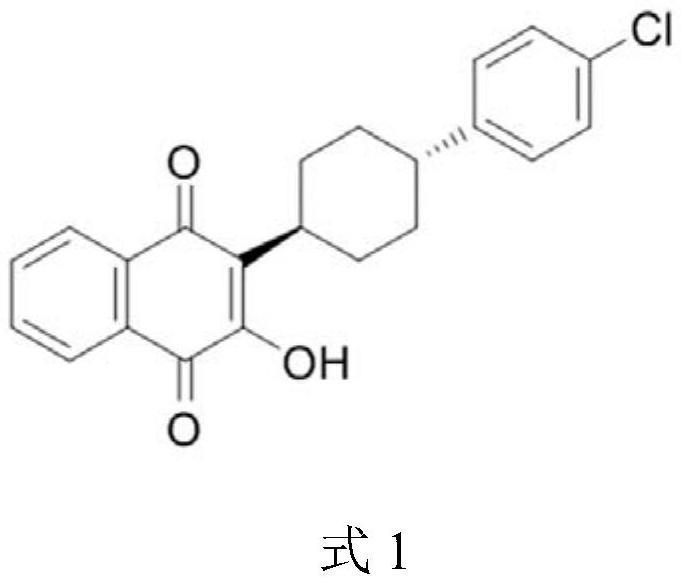
2、其中,所述阿托伐醌((3-[4-(4-氯苯基)环己基]-4-羟基萘-1,2-二酮))可以呈游离酸的形式(如式1所示),也可以呈药学上可接受的盐(如钠盐、钾盐)的形式。
3、
4、其中,所述伊维菌素为伊维菌素b1a、伊维菌素b1b(式2)或它们的混合物,优选含伊维菌素b1a和伊维菌素b1b的混合物,更优选伊维菌素b1a含量高于85%、伊维菌素b1a和伊维菌素b1b总含量高于90%的混合物。
5、
6、根据本发明,上述组合物对疟原虫红内期增殖、配子体形成的抑制具有显著协同增效作用,即组合物可获得比单一组分更好的抑制效果。
7、本发明也涉及在上述组合基础上进一步含有其它抗疟活性成分作为第三组分的组合物;
8、所述作为第三组分的其它抗疟活性成分包括,但不限于青蒿素、其衍生物或它们在药学上可接受的盐;
9、其中,所述青蒿素衍生物为二氢青蒿素、蒿甲醚或青蒿琥酯;
10、所述青蒿琥酯为二氢青蒿素半琥珀酸酯(式3)或其药学上可接受的盐(如钠盐、钾盐):
11、
12、本发明还涉及上述组合物用于抑制疟原虫增殖、配子体形成的用途,其具体施用方式可以是上述活性成分的同时施用或依次施用;
13、在活性成分同时施用的情况下,可将两种或三种活性成分组合在单独的药剂形式(固定组合,如单个药片或药囊等)之内。无论是否将活性成分同时、非同时、或部分同时(三种活性成分的情况下)施用,两种或三种活性成分可以以不同的药剂形式呈现。这种情况下,本发明所涉组合可以以组合包装药剂的形式呈现。
14、本发明还提供了基于在上述组合物的药物组合物,其进一步含有其它惰性组分或药学上可接受辅料,并进一步涉及所述组合物、药物组合物在制备疟疾治疗和/或预防药物中用途;
15、所述药物组合物可以以剂量单元的形式施用,每个剂量单元含有预定量的活性成分。取决于施用方法、途径,施用对象的患病状况、年龄、体重和健康情况,施用的剂量单元可包含不同剂量的本发明所涉组合。优选剂量单元配方,如日剂量或者份剂量,或是活性成分的相应比例,可由本领域的技术人员根据具体情况应用现有技术或者通过简单实验确定。这类药物组合物可以用药学领域已知的常用方法制备。
16、所述药物组合物可调整以适应于以任何需要的合适方式施用,例如经口(包括口腔或舌下)、经直肠、经鼻、局部(包括口腔、舌下或透皮)或胃肠外(包括皮下、肌肉内、静脉内或皮内)的方式。此类组合物可用药学领域已知的所有方法,如通过将活性成分与辅料或佐剂进行组合的方法制备。
17、适用于口服的药物组合物可以以独立的制剂单元施用,包括但不限于:胶囊或片剂;粉剂或颗粒;溶液、水或非水液体混悬液;可食用泡沫或泡沫食品;水包油液体乳或油包水液体乳。因此,以胶囊或片剂为例,活性成分组分可以与可口服、无毒且药学上可接受的惰性辅料,如乙醇、甘油和水等组合。通过将活性化合物粉碎至合适的细小尺寸并与以相同方式粉碎的药用辅料混合制备粉末,所述药用辅料,以可食用碳水化合物为例,可以是淀粉或甘露醇等。调味剂、防腐剂、分散剂和色素同样也可以用作辅料。
18、胶囊可通过上述方法制备粉末混合物并将其填充在成型的明胶胶囊壳中制备。在填充前,可在粉末混合物中加入诸如高分散硅酸、滑石粉、硬脂酸镁、硬脂酸钙或固态聚乙二醇等助流剂和润滑剂。为提高胶囊服用后药物的利用度,崩解剂或助溶剂,如琼脂、碳酸钙或碳酸钠等,也同样可以加入。此外,如有需要或必需,可在混合物中掺入合适的粘结剂、润滑剂和崩解剂以及色素。适用的粘结剂包括淀粉、明胶、天然糖(如葡萄糖或β-乳糖)、由玉米制得的甜味剂、天然或合成的增稠剂(如阿拉伯胶、黄芪胶或海藻酸钠、羧甲基纤维素、聚乙二醇、蜡类,等等)。制剂中使用的润滑剂包括油酸钠、硬脂酸钠、硬脂酸镁、苯甲酸钠、乙酸钠、氯化钠等。所述崩解剂包括但不限于淀粉、甲基纤维素、琼脂、膨润土、黄原胶等。
19、片剂可如下例配制:制备粉末混合物,经湿法或干法制粒,加入润滑剂和崩解剂,然后将其压制成片。粉末混合物的制备具体如下:将经适当方式粉碎的化合物与前文所述的药用辅料混合,并有选择地与粘结剂(例如羧甲基纤维素,海藻酸盐,明胶或聚乙烯吡咯烷酮)、溶出阻滞剂(如石蜡)、吸收促进剂(如四价盐)和/或吸附剂(例如膨润土,高岭土或磷酸二钙)混合。湿法制粒用粘结剂(如糖浆、淀粉糊、阿拉伯胶或纤维素/聚合物材料溶液)润湿粉末混合物,然后挤压过筛制粒。干法制粒直接将粉末混合物过压片机压制成形状不规则的块状物,然后将之粉碎成粒。可通过加入硬脂酸、硬脂酸盐、滑石粉或矿物油润滑制成的颗粒,以防止其粘结在压片模具上。然后将经润滑的颗粒压制成片。也可将活性成分与可自由流动的惰性辅料混合,然后直接压制成片,而无需先进行湿法或干法制粒。片剂可有虫胶密封包衣构成的透明或不透明的包含层,也可有糖或聚合物包衣和蜡打光包衣。可将色素加入到在上述包衣材料中以区分不同的剂量单元。
20、口服液,例如溶液、糖浆和酏剂,可以以剂量单元的形式制备,以便使给定份量含有预先确定量的化合物。糖浆可通过将化合物溶解于含合适矫味剂的水溶液中制备,酏剂则用非毒性醇溶媒制备。混悬液可通过将化合物分散于非毒性溶媒的方式配制。增溶剂和乳化剂(例如乙氧基化的异硬脂醇类和山梨醇聚氧乙烯醚类)、防腐剂、矫味剂(例如薄荷油、天然的甜味剂、糖精、或其它人造甜味剂等),同样可被加入到制剂中。
21、如有需要,可将经口施用的剂量单元配方包封于微胶囊中,也可以以释放延长或延迟的方式配制,例如通过将颗粒物包裹或包埋于聚合物、蜡等方式配制。
22、本发明所涉组合活性成分及其盐、溶剂化物和有生理功能的衍生物,以及其它活性成分也可以脂质体递送系统的方式施用,例如小单层囊泡、大单层囊泡和多层囊泡脂质体。脂质体可以由各种磷脂形成,如胆固醇、硬脂胺或磷脂酰胆碱。
23、适用于透皮施用的药物组合物可以以独立的贴剂单元施用,以与接受者的皮肤持续、紧密地接触。因此,本发明所涉组合的活性成分可以以药学领域所熟知的膏药形式施用,也可以如journal of pharmaceutical investigation.2021;51(5):503-517.中所概述的,以微针的形式施用。
24、适用于局部施用的药物组合物可以配制成软膏剂、乳膏剂、混悬剂、洗剂、粉剂、溶液剂、糊剂、凝胶剂、喷雾剂、气雾剂或油剂等。
25、适用于经直肠施用的药物组合物可以以栓剂或灌肠剂的形式施用。
26、适用于注射施用的溶液、混悬液可按配方用无菌粉末、颗粒和片剂制备。为达到活性成分的持续和/或可控释放,也可以如journal of controlled release.2017;267(10):57-66.中所概述的方法制备可注射水凝胶用于注射施用。
27、本发明提供的药物组合物及其制剂可作为人类和兽医学中的药物。
28、本发明所涉组合的每种活性成分和其它活性成分的治疗有效量或治疗活性剂量取决于多个因素,包括如患者/动物的年龄和体重,需治疗疾病的精准状况及其严重程度,处方制剂的特性和施用方式,最终需由主治医师或兽医确定。
29、对于含有阿托伐醌(游离酸)和伊维菌素(伊维菌素b1a含量高于85%、伊维菌素b1a和伊维菌素b1b总含量高于90%的混合物)为活性成分的本发明所涉组合,在经口施用情况下,活性成分的成人(假设体重为60kg)日剂量通常如下,阿托伐醌:0.5mg至4000mg,可优选100mg至3000mg,更优选500mg至2000mg或1000mg,每日可分1至4次施用;伊维菌素:6mg至120mg,可优选12mg至80mg,更优选24mg至60mg,每日可分1至4次施用。
30、对于以阿托伐醌(游离酸)、伊维菌素(伊维菌素b1a含量高于85%、伊维菌素b1a和伊维菌素b1b总含量高于90%的混合物)和青蒿琥酯为活性成分的本发明所涉组合,在经口施用情况下,活性成分的成人(假设体重为60kg)日剂量通常如下,阿托伐醌:0.5mg至4000mg,可优选100mg至3000mg,更优选500mg至2000mg或1000mg,每日可分1至4次施用;伊维菌素:6mg至120mg,可优选12mg至80mg,更优选24mg至60mg,每日可分1至4次施用;青蒿琥酯:2mg至200mg,可优选100mg至200mg,更优选150mg至180mg,每日单次施用。
31、基于以上,本发明还涉及一种用于治疗和/或预防疟疾的组合包装药品(由各活性组分独立制剂单元组成),首先包含阿托伐醌或其药学活性盐(第一活性组分),其次包含伊维菌素或其药学活性衍生物(第二活性组分)。所涉组合包装药品还可进一步包含青蒿素或其衍生物或药学上可接受的盐作为第三活性组分。
32、本发明所述的“有效量”指可在组织、系统、动物或人体引起施用者(例如研究者或医师)所寻求或期望的生物或医学反应的药物或药物活性成分的量。
33、本发明所述的“治疗有效量”指与未被施予该量的相应个体相比,具有以下结果的量:改善治疗,治愈,预防或消除疾病、综合征、病症、主诉、障碍或副反应,亦或减缓疾病、主诉或障碍的进展。“治疗有效量”的表述也包括可有效提升正常生理功能的量。
34、除另有说明外,此文所用的术语“前药”意指:活性成分的衍生物,其可在生物条件下(体外或体内)可通过水解、氧化亦或其它反应提供活性化合物。前药的范例包括但不限于带有生物可水解官能团的活性成分的衍生物和代谢物,诸如带有生物可水解酰胺、生物可水解酯、生物可水解氨基甲酸酯、生物可水解碳酸酯、生物可水解酰脲和生物可水解磷酸酯的类似物。前药通常可通过本领域已知的方法制备。
35、本发明具有如下有益技术效果:
36、减少单一活性组分的施用量,降低潜在毒副反应的风险;协同增效,提高总体施用对疟原虫增殖、配子体形成的抑制效果。
- 还没有人留言评论。精彩留言会获得点赞!