一种基于无规则空位信息的固有无序蛋白质预测方法
本发明涉及生物信息学,具体涉及一种基于多序列比对无规则空位信息的固有无序蛋白质预测方法。
背景技术:
1、蛋白质是生命活动的直接执行者,其结构和动力学性质与生理功能密切相关。早期建立在大量晶体结构基础上的蛋白质研究认为稳定的三维结构是蛋白质行使生物学功能所必需的,即“序列-结构-功能”的范式:蛋白质的氨基酸序列决定三维结构,三维结构则决定其生物学功能。随着结构生物学和生物物理化学的发展,人们发现大量蛋白质在行使功能时会发生构象变化,并存在多种不同的构象。这些研究表明蛋白质的动力学性质与其功能密切相关,推动蛋白质研究进入“结构-动力学-功能”时代。
2、21世纪以来,人们开始注意到很多在天然状态下没有稳定三维结构的蛋白质或蛋白质区域依然发挥重要的生物学功能。这类在天然状态下不具有稳定三维结构的蛋白质或区域被称为固有无序蛋白质/固有无序蛋白区域。在高等生物中,固有无序蛋白质/固有无序蛋白区域的含量明显高于低等生物。在人类基因组中,约40%的蛋白质含有长度大于30个氨基酸的无序区域。固有无序蛋白质与多种疾病密切相关,例如,79%的癌症相关蛋白含有长度大于30个氨基酸的无序片段。因此,固有无序蛋白质是一类重要的药物靶标,相关研究为药物研发提供了新的机遇。然而,由于大多数理性药物设计策略都依赖于高分辨率的蛋白质三维结构,固有无序蛋白质所具有的动态特性给相关药物设计造成了巨大的障碍。因此准确识别固有无序蛋白质对研究蛋白质功能以及设计药物具有重要的意义。
3、基于传统的实验技术识别固有无序蛋白质具有周期长耗费大的缺点,因此开发快速准确的计算方法变得尤为重要。现有的计算方法可以分为四类:基于物理化学性质的方法、基于机器学习的方法、基于模板的方法和组合分类器。蛋白质的有序区域具有稳定的三维结构并且具有高的序列保守性,与无序区域相比更容易生成高质量序列比对。而现有的计算方法缺乏对两者之间差异更直观的描述,导致对固有无序蛋白质的预测性能不佳。
4、因此,有必要发明一种基于多序列比对无规则空位信息的固有无序蛋白质预测方法,以解决上述问题。
技术实现思路
1、本发明的目的在于提供一种预测性能佳、成本低、可快速准确地识别固有无序蛋白质的预测方法。
2、为达到上述目的,本发明提供如下技术方案:
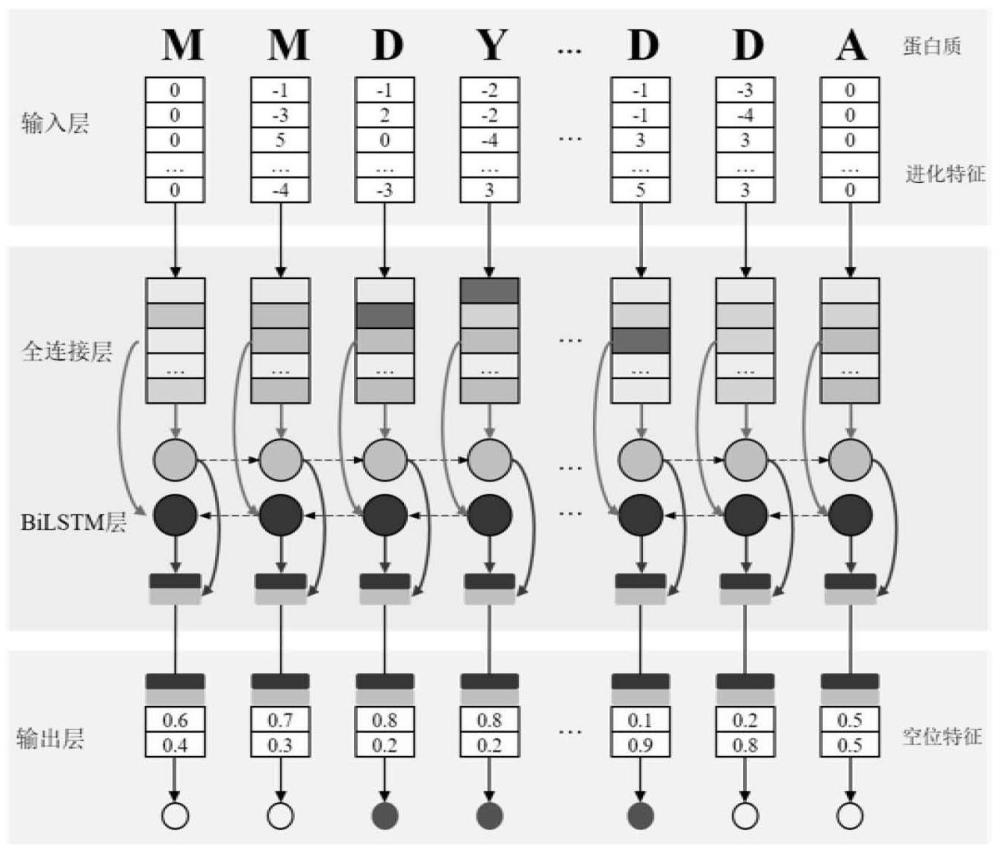
3、一种基于多序列比对无规则空位信息的固有无序蛋白质预测方法,其特征在于,包括如下步骤:
4、一种基于多序列比对无规则空位信息的固有无序蛋白质预测方法,其特征在于,包括如下步骤:
5、s1、搜索蛋白质数据库,构建待预测的固有无序蛋白质的多序列对比信息;
6、所述多序列比对信息中,位置i上的氨基酸残基的空位特征为:
7、
8、其中,和分别代表所述多序列比对信息的位置i上空位的个数和非空位的个数,每个氨基酸残基的空位特征为[msagi,1-msagi];
9、s2、利用所述多序列对比信息,生成所述固有无序蛋白质的位置特异性得分矩阵pssm;
10、s3、构建固有无序蛋白质预测方法idp-msa的模型,所述模型包含输入层、全连接网络层、双向长短时记忆网络层和输出层,所述模型的工作原理为:
11、通过所述输入层,将蛋白质序列表示为特征向量;
12、通过所述全连接网络层学习输入的所述蛋白质序列的氨基酸残基特征之间的线性关系,丰富所述蛋白质序列的氨基酸残基特征,并浓缩所述特征向量的维度;
13、通过所述双向长短时记忆网络层学习所述蛋白质序列的氨基酸残基特征的局部上下文特征及远距离的依存关系,利用全连接网络和双向长短时记忆网络学习所述蛋白质序列的进化特征潜在的特征表示,得到潜在特征;
14、将所述空位特征及所述潜在特征输入所述输出层进行处理,采用sigmoid函数进行分类,输出每个氨基酸残基属于无序和有序的概率值;
15、s4、对训练集执行步骤s3,对所述模型进行训练;
16、s5、对测试集执行步骤s1、s2后将pssm输入所述模型,执行步骤s3,得到预测结果。
17、进一步地,s1步骤中,采用psi-blast和hhblits两种方法构建蛋白质的多序列比对信息,分别记为bmsa和hmsa,并将两种方法提取的空位特征分别表示为bmsag和hmsag。
18、进一步地,s4步骤中,使用adam优化算法对所述模型进行训练。
19、优选地,s4步骤中,使用概率为0.6~0.8的dropout层对所述模型进行训练;更优选地,使用概率为0.7的dropout层对所述模型进行训练.。
20、进一步地,s4步骤中,选择带有权重的交叉熵函数作为训练过程中的损失函数。
21、本发明的有益效果在于:
22、本发明基于固有无序蛋白质的多序列比对信息,构建了固有无序蛋白质的有序区域与无序区域在进化中表现出的无规则空位特征,并结合双向长短时记忆网络构建了固有无序蛋白质预测方法idp-msa;本发明提出的预测方法不仅能够学习蛋白质序列中氨基酸残基的局部上下文特征和远距离的依存关系,而且能够更直接地刻画有序区域和无序区域在序列保守性上的差异性,大幅度提高了固有无序蛋白质的预测性能;此方法可快速准确地识别固有无序蛋白质,成本低,可行性强,便于使用和推广。
23、上述说明仅是本发明技术方案的概述,为了能够更清楚了解本发明的技术手段,并可依照说明书的内容予以实施,以下以本发明的较佳实施例并配合附图详细说明如后。
技术特征:
1.一种基于多序列比对无规则空位信息的固有无序蛋白质预测方法,其特征在于,包括如下步骤:
2.如权利要求1所述的基于多序列比对无规则空位信息的固有无序蛋白质预测方法,s1步骤中,采用psi-blast和hhblits两种方法构建蛋白质的多序列比对信息,分别记为bmsa和hmsa,并将两种方法提取的空位特征分别表示为bmsag和hmsag。
3.如权利要求1所述的基于多序列比对无规则空位信息的固有无序蛋白质预测方法,s4步骤中,使用adam优化算法对所述模型进行训练。
4.如权利要求1所述的基于多序列比对无规则空位信息的固有无序蛋白质预测方法,s4步骤中,使用概率为0.6~0.8的dropout层对所述模型进行训练。
5.如权利要求1所述的基于多序列比对无规则空位信息的固有无序蛋白质预测方法,s4步骤中,选择带有权重的交叉熵函数作为训练过程中的损失函数。
技术总结
本发明涉及一种基于多序列比对无规则空位信息的固有无序蛋白质预测方法。本发明基于固有无序蛋白质的多序列比对信息,构建了固有无序蛋白质的有序区域与无序区域在进化中表现出的无规则空位特征,并结合双向长短时记忆网络构建了固有无序蛋白质预测方法IDP‑MSA。本发明提出的预测方法不仅能够学习蛋白质序列中氨基酸残基的局部上下文特征和远距离的依存关系,而且能够更直接地刻画有序区域和无序区域在序列保守性上的差异性,大幅度提高了固有无序蛋白质的预测性能;此方法可快速准确地识别固有无序蛋白质,成本低,可行性强,便于使用和推广。
技术研发人员:刘羽朦,靳小鹏,谢俊熙
受保护的技术使用者:深圳技术大学
技术研发日:
技术公布日:2024/1/15
- 还没有人留言评论。精彩留言会获得点赞!