多模式注释生成的基因突变预测方法与流程
本发明涉及基因突变预测,具体是多模式注释生成的基因突变预测方法。
背景技术:
1、全基因组测序(wgs)和全外显子组测序(wes)数据的快速积累导致了大量病理性和非病理性遗传突变的发现。为了帮助评估和理解这些突变,世界上已经有研究机构建立了人群数据库,如gnomad、exac和chinamap等。此外,遗传疾病数据库,如clinvar、omim、hgmd,也积累了大量已知病理性或良性遗传突变的信息。这些数据库已被广泛用作孟德尔遗传病的遗传诊断参考。
2、已知导致孟德尔遗传病的病理性突变通过各种生物学机制发挥作用,因此在不同方面有广泛的分类和研究。例如,基于蛋白质序列改变的外显子突变被分类为同义突变、错义突变、终止突变、终止缺失、移码突变等。同义突变不改变蛋白质序列,而错义突变导致编码不同的氨基酸。由于与错义突变相关的蛋白质序列变化可能具有病理性,各种研究已经集中于预测错义突变的病理影响。另一方面,一些突变在rna水平上通过剪接改变具有病理性,这些突变通常位于剪接供体、受体和内含子区域。因此,剪接突变也被考虑用于突变的病理评估。然而,在使用全外显子测序数据进行实际遗传诊断时,应同时考虑不同类型的突变和机制,以识别病理性突变。随着机器学习(ml)和深度学习(dl)的发展,许多使用ml或dl的计算方法已被开发用于预测突变破坏或病理性。
3、上述领域中一些算法考虑从多重维度获得的集成特征,并在现有致病性预测的基础上构建。例如,mutpred2(mutation predictor 2,突变预测器)[1]和revel(rare exomevariant ensemble learner,罕见外显子突变集成学习)[2]。mutpred2的输入是一个氨基酸序列s,即一个野生型蛋白质序列,和一个氨基酸替换xiy,其中x是s中的第i个氨基酸,被y替换。我们将突变(mt)序列称为sxiy。mutpred2使用hgmd(human gene mutation database,人类基因突变数据库),swissprot,dbsnp作为训练集。对于给定的一个序列s和变体xiy,mutpred2提取了1345个特征(包括20个可选特征)。这些特征被分成六组:(1)基于序列的特征,(2)基于氨基酸替代的特征,(3)基于pssm(position-specific scoring matrix,位置特异性评分矩阵)的特征,(4)基于保守性的特征,(5)同源蛋白质特征(由于计算时间需要,可选),以及(6)预测结构和功能性质的变化。mutpred2使用双样本t检验进行特征选择,只保留返回p值<0.01的特征。为了去除(近似)共线性特征,对所选特征进行z-score标准化和主成分分析,保留方差设置为99%。然后,在得到的特征矩阵上训练了30个前馈神经网络的集合。每个网络由一个具有四个神经元和一个输出神经元的隐藏层组成(两个层都使用tanh激活函数)。采用bagging方法进行训练,每个网络在原始训练集的平衡随机样本上有放回地进行训练。为了确定训练所需的迭代次数,mutpred2将25%的训练数据保留为验证集。最终模型使用resilient propagation算法进行训练,并在达到最佳迭代次数、完成1000轮或达到500个检查点时停止。然后计算预测得分为所有30个验证检查输出得分的平均值。mutpred2的输出包括一个取值范围在[0,1]之间的致病性评分,以及可能受到xiy影响的分子机制评分列表。致病性评分为1表示突变几乎肯定是致病的,而评分为0表示突变几乎肯定是良性的。
4、revel是nilah等人提出的一种方法。revel的训练集来自hgmd,esp(exomesequencing project,外显子测序计划)和kgp(1000genomes project,千人基因组计划)。revel将来自13个工具的18个致病性预测分数作为预测特征进行整合。其中包括10个功能预测分数(mutpred、provean、sift、polyphen-2 hvar&hdiv、lrt、mutationtaster、mutationassessor、fathmm v2.3和vest 3.0)以及8个保守性分数(gerp++、siphy、灵长类、脊椎动物、哺乳动物的phylop和phastcons分数)。revel对于缺失的特征值使用r软件包中实现的k最近邻方法进行插补。对于给定的突变,缺失的特征值被赋予其k个最近邻突变的非缺失特征值的平均值;当给定突变的超过50%的特征值缺失时,将每个缺失的特征值赋予其在所有突变中的整体平均值。最后revel使用包含1,000个二元分类树的随机森林算法进行训练
5、尽管现有的突变致病性预测算法被广泛使用,采用了截止各自发表时最先进的技术进行开发,但它们大多只适用于特定类型突变或依赖于已经发表的突变致病性预测工具的分数作为先验知识,在实际预测任务中有某些类型遗传突变的致病性无法预测。
技术实现思路
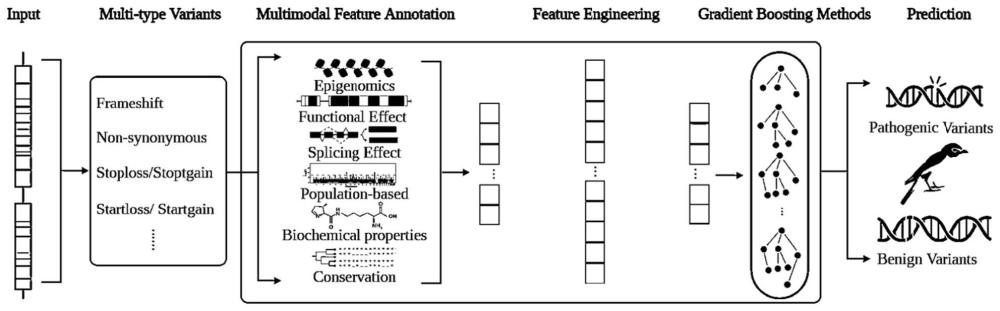
1、本发明要解决的技术问题是提供多模式注释生成的基因突变预测方法,用于预测所有非同义外显子突变。
2、为解决上述技术问题,本发明提供多模式注释生成的基因突变预测方法,过程包括将输入的单碱基突变位置信息进行突变类型注释获得包含突变种类的突变基本信息,然后进行多维特征注释获得包含注释结果的多维特征的突变数据集,再使用基于贝叶斯pca对包含注释结果的多维特征的突变数据集进行填补注释数据的操作,然后使用自动工程特征列表对基于贝叶斯pca填补的注释数据进行特征生成,再使用分离特征选择列表进行数据筛选,将所获得的包含所有筛选特征后的突变数据集经过梯度生成树算法后获得基因突变的预测分数。
3、作为本发明的多模式注释生成的基因突变预测方法的改进:
4、所述突变类型注释为使用refgene数据库进行进行突变类型注释;
5、所述多为特征注释包括使用annovar注释工具、spliceai剪接效应预测软件、功能效应数据库参考突变信息进行注释。
6、作为本发明的多模式注释生成的基因突变预测方法的进一步改进:
7、所述annovar注释工具为对于基于人群的特征、氨基酸的生化特性改变特征和保守性分数特征进行注释:
8、对于基于人群的特征,包括检索各种人群中的等位基因频率:全外显子(af)、原始等位基因频率(af_raw)、非洲人(af_afr)、拉丁美洲人/混血美洲人(af_amr)、阿什肯纳兹犹太人(af_asj)、东亚人(af_eas)、芬兰人(af_fin)、非芬兰欧洲人(af_nfe)和其他人群(af_oth),还从注释信息中获取不同性别的等位基因频率;
9、对于氨基酸的生化特性改变特征首先检查突变是否导致氨基酸变化,如果没有,将所有相关特征设为0;
10、将每个氨基酸的物理化学性质存储在一个矩阵中,通过查询矩阵获取氨基酸的相应属性,并将突变前后的属性差作为突变的特征;当一个突变影响多个氨基酸时,分别计算变化前后的平均值;还使用从blosum100矩阵中获得的信息作为特征;
11、保守性分数特征包括在灵长类动物、哺乳动物、脊椎动物中的phastcons、phylop和siphy的分数特征。
12、作为本发明的多模式注释生成的基因突变预测方法的进一步改进:
13、所述spliceai剪接效应预测软件为对于剪接效应特征进行注释获得每个突变体对剪接的预测效果和与剪接变化相对于突变体位置的信息;
14、采用突变体的改变分数表示突变体对剪接的影响概率,包括受体增益(ds_ag)、受体丧失(ds_al)、供体增益(ds_dg)和供体丧失(ds_dl),与剪接变化相对于突变体位置的信息包括受体增益(dp_ag)、受体丧失(dp_al)、供体增益(dp_dg)和供体丧失(dp_dl)的位置差;
15、如果spliceai不对突变体进行注释,则表示突变体靠近染色体末端或参考序列过长使用0来填充spliceai预测的缺失值。
16、作为本发明的多模式注释生成的基因突变预测方法的进一步改进:
17、所述功能效应数据库为对于表观基因组学特征和基因损伤指数(gdi)、残余突变不耐受性评分(rvis)、基于功能丧失工具(loftool)的基因不耐受性评分(lof_score)和omim的功能效应特征,使用功能效应数据库对照突变位置进行注释:
18、每个突变的表观基因组学特征由15状态的chromhmm模型在九种不同细胞系中进行注释,以捕捉不同染色质标记之间的相互作用(染色质状态)的空间上下文;
19、功能效应包括基因损伤指数(gdi)、残余突变不耐受性评分(rvis)、基于功能丧失工具(loftool)的基因不耐受性评分(lof_score)、突变体类型以及来自omim数据库的注释;使用突变类型作为特征,包括错义突变、终止突变、起始缺失突变、移码突变、非移码突变和终止缺失突变;
20、使用omim数据库对突变遗传模式进行注释,将其分为五种不同类型,包括常染色体隐性、常染色体显性、x连锁隐性、x连锁显性和其他;
21、然后对于表观基因组学特征、突变体类型特征和突变遗传模式特征进行one-hot编码。
22、作为本发明的多模式注释生成的基因突变预测方法的进一步改进:
23、所述梯度生成树算法包括lgbm模型,lgbm模型训练过程为:构建训练集和测试集,然后经过突变类型注释、多维特征注释、带有分离特征选择的自动特征工程处理后获得包含选择后所有特征的训练集和测试集,对lgbm模型采用5轮训练,每轮均使用5折交叉验证和逐步参数调整,从而获得训练好的lgbm模型。
24、作为本发明的多模式注释生成的基因突变预测方法的进一步改进:
25、所述带有分离特征选择的自动特征工程的过程具体为:
26、(1)使用基于贝叶斯pca的缺失值估计方法对包含多维特征的突变数据集填补特征的缺失值;
27、(2)自动特征工程:
28、采用openfe软件包执行,对原始特征进行对数、平方根和指数等数学变换的操作并根据特征的值将原始特征分组生成新的分类特征,其次,使用openfe中定义的默认方法进行特征选择所获得的特征保存为所述自动工程特征列表;
29、(3)分离特征选择:
30、对于自动特征工程获得的特征的数据集,使用lgbm模型进行不超过50轮的特征选择以将特征数量减少到200个或更少,在每一轮特征选择之后,评估每个特征的重要性,并丢弃相对重要性得分低于1e-3的特征;然后将经过50轮筛选后的特征分为两个类别,即核心特征集和附加特征集;使用分离特征选择算法中输出的特征组合保存为所述分离特征选择列表。
31、作为本发明的多模式注释生成的基因突变预测方法的进一步改进:
32、所述分离特征选择算法的具体过程包括:
33、输入:核心特征集c={c1,c2,…,cn},附加特征集a={a1,a2,…,an},最终特征数目m;
34、
35、输出:有最优性能的特征组合{c,ak}。
36、作为本发明的多模式注释生成的基因突变预测方法的进一步改进:
37、所述训练集和测试集具体构建的过程为:
38、获取clinvar数据库并过滤删除了冲突标签和未知标签的突变;
39、获取gnomad数据库并选择了等位基因频率在1e-5和1e-3之间的罕见良性突变并进行筛选处理:在每个染色体中随机选择5,000个突变,通过annovar注释后过滤掉用于突变预测特征中存在缺失的突变,然后再次随机选择保留每个染色体的500个突变;chr11和chry染色体保留全部突变并随机使用其他染色体中的合格突变填补空缺;
40、获取并筛选出唯一reference snp id对应的突变从而构建了正交验证集swissprot;
41、按1.644:1的比例将gnomad库的突变分为两个子集gnomadclinvar和gnomadswissprot,然后将gnomadclinvar与clinvar合并为clinvargnomad,将gnomadswissprot和swissprot合并为swissprotgnomad;使用拆分算法将clinvargnomad拆分获得划分后的数据集a和数据集b,数据集s和数据集b分别作为训练集和测试集。
42、作为本发明的多模式注释生成的基因突变预测方法的进一步改进:
43、所述拆分算法的具体过程为:
44、(1)、初始化两个空数据集a、b;两个基因列表la=[]、lb=[];a在d中的实时比例na;
45、(2)、while true
46、(3)、do ifna≥n
47、(4)、then terminate and return{a,b}
48、(5)、endif
49、(6)、从ld中随机选择一个基因g,ng为d中该基因对应突变数量
50、(7)、la=la∪g,ld=ld-g,na+=ng
51、(8)、endwhile
52、(9)、lb=ld
53、(10)、a=d[gene inla],b=d[gene inlb]
54、(11)、returna,b
55、其中,d为待划分的数据集,la、lb和ld为包含数据集a、b和d的基因列表,n为应添加到a中的突变比例阈值为0.9,na为算法运行过程中a占d的实时比例。
56、本发明的有益效果主要体现在:
57、1、本发明通过添加了来自gnomad超过10,000个罕见良性突变(allele frequency(af,突变等位基因频率)在0.00001到0.0001之间)用于模型的训练,以提高模型在分类罕见良性突变方面的性能,这在减少假阳性率方面至关重要;
58、2、本发明的magpie模型提取了6种特征模态,包含具有较少缺失率的特征,从而扩大可预测的突变类型范围,并对数据集中的特征采用基于bayes-based principalcomponent analysis(bpca,基于贝叶斯的主成分分析)的方法进行填补;采用带有分离特征选择的自动特征工程,根据特征的特点和训练数据集,从而从数据集中提取了尽可能多的信息;
59、3、本发明的magpie模型中应用了基于梯度生成树算法的light gbm模型(简称为lgbm模型,light gradient boosting machine)模型,并与其他使用机器学习或深度学习模型的工具相比取得了更好的性能,magpie能够对多种类型的外显子突变进行预测,填补了以前方法中不适用的缺失值的5-60%;magpie在高度不平衡的验证数据集以及低人群等位基因频率的突变中表现出较好的性能,突出了在个体患者的临床相关应用中解释未知致病性突变的优势,可以用于从大量候选突变中识别少量致病概率较高突变。
- 还没有人留言评论。精彩留言会获得点赞!