一种反钙钛矿衍生材料设计与高通量模拟方法
本发明涉及反钙钛矿半导体,尤其是涉及一种反钙钛矿衍生材料设计与高通量模拟方法。
背景技术:
1、能源危机和气候变暖是全球面临的两大严峻问题,近年来,钙钛矿太阳能电池凭借其制备工艺简单、原材料成本低廉、光电转换效率高等优势,吸引了全球科学家们的广泛关注。目前,钙钛矿太阳能电池的最高光电转换效率(pce)已经超过了25%,接近商用单晶硅太阳能电池的效率。但这些高效钙钛矿器件的核心吸光层材料多基于含铅卤化物钙钛矿,而铅(pb)的毒性问题给光伏器件的商业化带来了重要挑战。设计合成新型低毒甚至无毒的高效无铅光伏材料,既能降低环境毒性,又具有重要的科学意义和商业前景,是未来新一代光伏技术发展的主要趋势。
2、为了解决铅的毒性问题,最直接有效的方法是用同族或者相邻族金属阳离子取代pb2+,即设计合成无铅钙钛矿光伏材料。目前,研究者已尝试使用各种具有相似ns2电子构型的金属阳离子来取代pb2+,如sn2+(5s25p0)、ge2+(4s24p0)、sb3+(5s25p0)、bi3+(6s26p0)等。尽管无铅钙钛矿光伏材料能够有效降低环境毒性,但是存在较大的带隙值、差的载流子传输性能、深的缺陷能级等问题,这导致实验报道的无铅钙钛矿器件依然面临光电转换效率低(<10%)等严峻的挑战,难以达到商业化的要求。因此,亟需发展新的策略来设计高效无铅光伏材料。
3、近年来,一些研究者开始探索将反钙钛矿半导体材料应用在光伏领域。反钙钛矿结构不仅给新型光电半导体领域提供了新的材料设计平台,而且为开发无毒高效太阳能电池材料提供了全新的解决思路,具有重要的潜在应用前景。
4、在有机无机杂化反钙钛矿的光电性能研究方面:美国andrew m.rappe课题组在2017年首次从理论层面预测了一系列有机无机杂化反钙钛矿化合物(ma)3ba。这些化合物中,b位和a位被-2或-1价的阴离子所占据。通过第一性原理计算,他们揭示了这些预测的化合物具有广泛可调的带隙。考虑到有机阳离子位置的变化(例如,从a位变为x位),他们提出反钙钛矿结构可能有望克服目前有机无机杂化卤化物钙钛矿太阳能电池在稳定性方面所面临的问题。接着,他们通过对b位和a位进行不同组分的替代,预测了一系列新的有机无机杂化反钙钛矿体系。然而,遗憾的是,这些理论上预测的有机无机杂化反钙钛矿化合物具有较平的导带底和价带顶,意味着这些材料体系具有非常大的载流子有效质量,不利于作为单节太阳能电池的吸光层材料。
5、在无机反钙钛矿的基态结构和光电性能研究方面:1992年,美国科学家disalvo等人首次在实验中合成了无机反钙钛矿ca3mn(m3-=p,as,sb,bi),并提供了其基本的晶体结构信息。然而,直到近年来,日本的研究人员才利用第一性原理计算工具从理论上对这些材料的基态晶体结构进行了深入研究,这些不同组分之间由于离子半径的差异,导致它们在基态下拥有不同的空间群。
6、同时,研究人员还对这些材料的光电性能进行了调查,发现其中ca3bin、ca3sbn、mg3pn和sr3pn可能具有作为单节太阳能电池吸光层材料的潜力。最近,华中科技大学的研究团队成功地合成了四种反双钙钛矿体系,分别是li6nbrbr2、li6nii2、li6nclbr2和li6nbri2。然而,这些材料都具有非常大的间接带隙值(>3.5ev),因此不适合用作光伏材料。
7、总体来说,目前具有光伏应用潜力的高性能反钙钛矿半导体的组分空间还有待进一步开拓,这些材料的高效光电性能微观机制以及构效关系有待深入理解。
技术实现思路
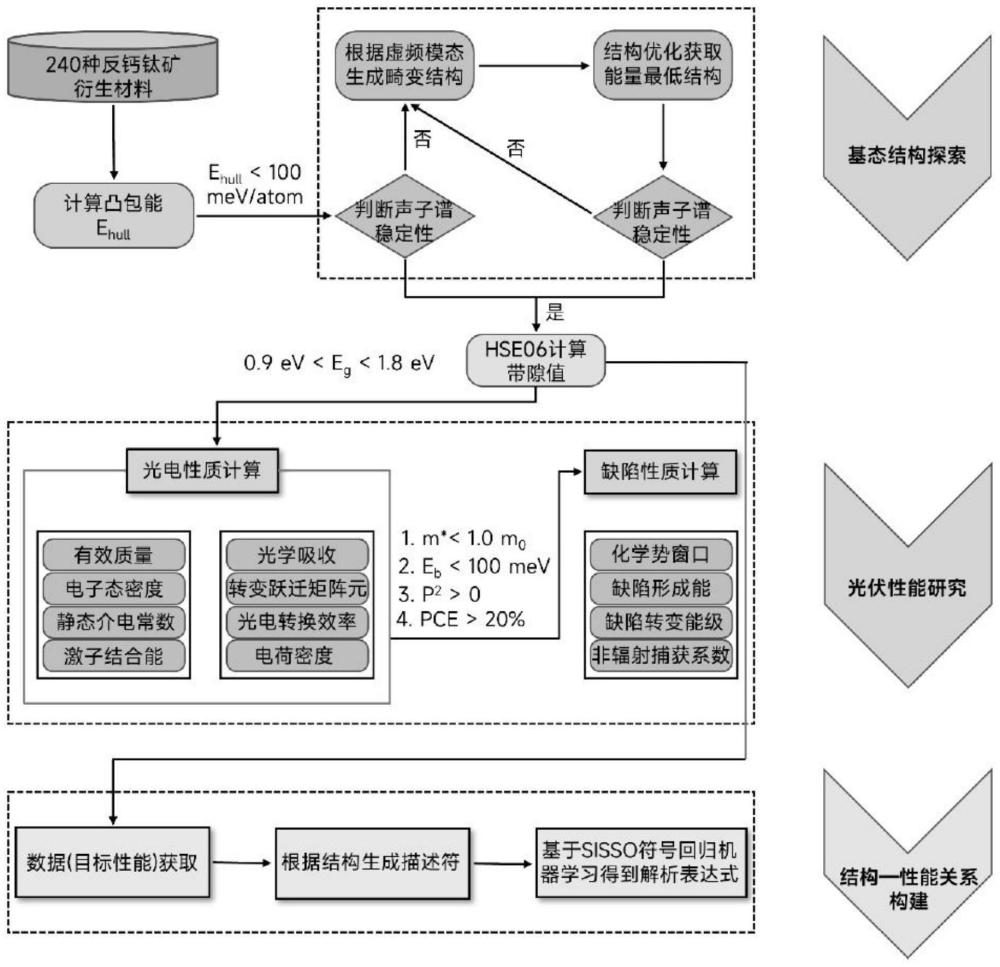
1、本发明的目的是提供一种反钙钛矿衍生材料设计与高通量模拟方法,基于功能基元思想开发了原子坐标劈裂设计策略,旨在设计全新的无铅反钙钛矿衍生材料;并结合第一性原理晶格动力学和群论方法探究新的无铅反钙钛矿衍生材料相竞争机制。
2、为实现上述目的,本发明提供了一种反钙钛矿衍生材料设计和高通量模拟方法,包括如下步骤:
3、s1、基于wyckoff位置劈裂策略,将反钙钛矿x3ba中的a位原子位置(0,0,0)劈裂为3个a′位置(0.5,0,0)、(0,0.5,0)和(0,0,0.5),获得反钙钛矿衍生材料x3ba′3;
4、s2、在x3ba′3的基础上应用b位阴离子序策略,将[x6b]八面体变换成有序排列的[x6b′]和[x6b″]八面体,获得反钙钛矿衍生材料x6b′b″a′6;
5、s3、构建240种x3ba′3和x6b′b″a′6反钙钛矿衍生材料的初始晶体结构文件,其中,x3ba′3空间群为pm-3m,x6b′b″a′6空间群为fm-3m;之后基于第一性原理程序筛选得到热力学和动力学稳定的候选材料;
6、s4、计算热力学和动力学稳定的候选材料的带隙eg,选择带隙在0.9ev<eg<1.8ev的热力学和动力学稳定的候选材料为候选材料ⅰ;验证候选材料ⅰ带隙的准确性,对验证无误的候选材料ⅰ进行光电性质和缺陷物理的研究;
7、s5、基于机器学习技术构建240种x3ba′3和x6b′b″a′6反钙钛矿衍生材料的结构—性能关系。
8、所述步骤s3筛选流程包括:
9、s3-1、选择初始晶体结构文件中的一种x6b′b″a′6结构,构建其[x6b′]和[x6b″]八面体不同排列方式的模型,并进行结构优化确定能量最低的构型;
10、s3-2、根据步骤s3-1中获得的能量最低的构型,对240种x3ba′3和x6b′b″a′6进行结构优化,并获取优化后的能量值;
11、s3-3、结合优化后的能量值和materials project数据库中所有竞争相的能量值,计算240种x3ba′3和x6b′b″a′6的凸包能ehull,并选择其中ehull<100mev/atom的材料作为热力学稳定候选材料;
12、s3-4、计算热力学稳定候选材料的二阶力常数,并绘制其声子色散曲线,声子谱在任意点均不出现虚频的为动力学稳定的候选材料;
13、s3-5对于声子谱存在虚频的热力学稳定候选材料,根据虚频模态生成畸变结构,对畸变结构进行结构优化确定能量最低结构,基于能量最低结构继续进行声子谱计算,重复以上流程至声子谱在任意点均不出现虚频,此晶体结构即为动力学稳定的候选材料。
14、优选的,所述步骤s4进行候选材料ⅰ光电性质和缺陷物理的研究包括:
15、s4-1、借助第一性原理程序进行候选材料ⅰ的光电性质计算并筛选出优异光电性能的候选材料ⅱ;
16、s4-2、所述光电性质计算包括:电子me和空穴有效质量mh、带隙eg、静态介电常数εstd、激子结合能eb、光学吸收α、转变跃迁矩阵元p、光电转换效率pce;
17、s4-3、所述筛选标准为:me/mh<1.0m0,其中m0是电子的静态质量、0.8ev<eg<1.6ev、εstd>20、eb<100mev、α>10-5cm-1、p2>0、pce>20%;
18、s4-4、采用第一性原理程序计算候选材料ⅱ的缺陷性质,筛选具备缺陷容忍特性的候选材料ⅲ。
19、优选的,所述步骤s5包括:
20、s5-1、以步骤s4中的带隙eg作为初始输入,借助python工具包构建机器学习的数据集;
21、s5-2、借助开源软件matminer从数据集中的化学式构建描述符;
22、s5-3、通过皮尔逊相关系数方法,判断描述符和带隙eg之间的相关性,筛选出和带隙eg之间皮尔逊相关系数>0.9的描述符ⅰ;
23、s5-4、将描述符ⅰ作为机器学习的训练集,采用开源软件sisso进行符号回归机器学习,选择出决定系数r2接近1、均方根误差rmes较小的模型,并根据其绘制240种x3ba′3和x6b′b″a′6反钙钛矿衍生材料的结构—性能关系图。
24、优选的,所述步骤s1中x=mg,ca,sr,ba;b=n,p,as,sb,bi;a′=f,cl,br,i。
25、优选的,所述步骤s2中x=mg,ca,sr,ba;b′b″=np,nas,nsb,nbi,pas,psb,pbi,assb,asbi,sbbi;a′=f,cl,br,i。
26、优选的,对于所述步骤s3-5中从高对称相演变到低对称相的畸变结构,借助群论工具进行分析,确定反钙钛矿衍生材料的晶体结构演化规律。
27、因此,本发明采用上述一种反钙钛矿衍生材料设计与高通量模拟方法,具有如下技术效果:
28、(1)基于功能基元思想提出原子位置劈裂设计策略,然后结合阴离子序策略,从反钙钛矿结构衍生出两类新型类反钙钛矿结构半导体材料;解决了传统的原子种类取代策略所设计出的无铅光伏材料往往存在光电转换效率低的问题;
29、(2)借助第一性原理晶格动力学和群论方法,从立方高对称相的声子谱曲线出发,根据虚频振动模态生成低对称畸变结构,并借助群论工具探究从高对称相演变到低对称相的规律,最终揭示反钙钛矿衍生材料的晶体结构演化规律;
30、(3)采用最新的机器学习技术,通过回归模型来探究反钙钛矿衍生材料的带隙等性能和结构参数之间的关联,确立其构效关系模型。
31、下面通过附图和实施例,对本发明的技术方案做进一步的详细描述。
- 还没有人留言评论。精彩留言会获得点赞!