一种用于加速第三代RNA测序数据比对的最大精确匹配并行查找算法
本发明涉及生物信息学的序列比对加速算法,具体是一种用于加速第三代rna测序数据比对的最大精确匹配并行查找算法。
背景技术:
1、随着第三代测序技术的崭露头角,基因组学领域正经历着一场历史性的变革。与前两代测序技术不同,第三代测序技术通过生成更长的读段,它能够跨越整个转录本,使得研究人员能够解读更为复杂的基因结构,并深入研究复杂的可变剪接事件。同时,基因组领域正在迎来后基因组时代,其特征在于通过全面的基因组测序以更好地理解基因表达与调控的微妙差异。
2、其中,rna测序对于揭示基因表达的复杂性具有深远的影响。后基因组时代的兴起为揭示复杂的基因调控机制提供了强大的动力,并推动了rna测序数据分析维度的深刻重构。随着大量rna测序数据比对算法的深入研究,当前的算法在准确性方面取得了显著的进展,进而推动了rna测序的广泛应用,直接导致测序数据的激增。这一激增开启了基因组学的大数据时代,对算法的速度提出了新的挑战,以更快、更有效地处理海量数据。现代计算机通常具备多核能力,因此研究如何充分利用cpu以加速比对算法,对于加速rna测序数据的分析具有重要的现实意义。
技术实现思路
1、本发明的目的在于加速第三代rna测序数据比对,为此提供了一种并行的最大精确匹配查找算法,该算法具有运行速度快、内存消耗低和数据吞吐量高的特点,能够有效的加速比对过程,从而有助于rna测序数据的快速分析。
2、实现本发明的技术方案为:
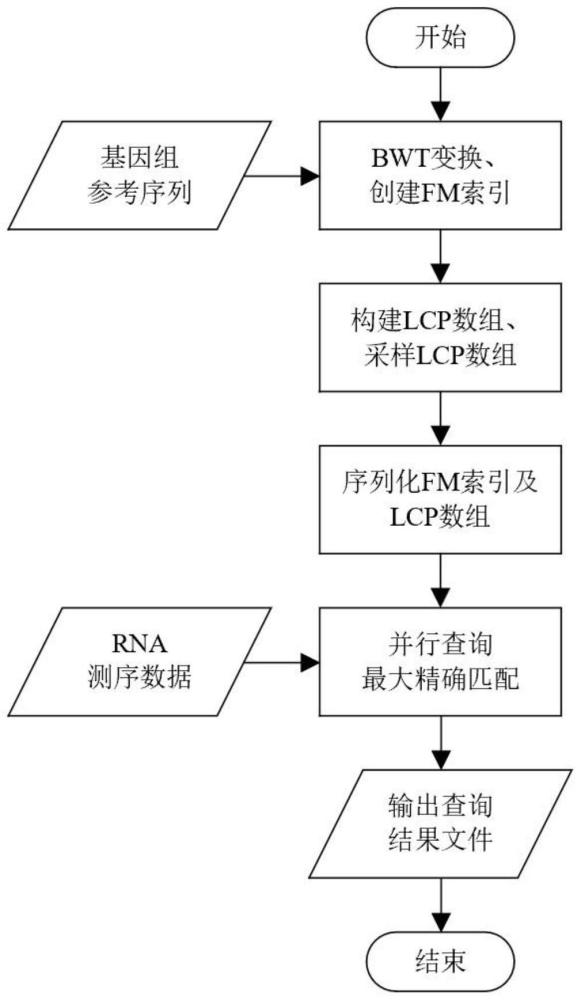
3、一种用于加速第三代rna测序数据比对的最大精确匹配并行查找算法,包括以下步骤:
4、(1)收集数据集,包括3个真实数据集和1个模拟数据集,每个数据集包括参考基因组的查询序列ref,以及第三代rna测序数据reads;
5、(2)对参考基因组序列ref进行bwt变换,其中,bwt变换为burrows-wheelertransform变换,它对参考序列的所有轮转字符串进行排序,得到轮转字符串数组,这样可以将相似的字符聚集在一起;
6、(3)构建参考序列的fm索引,其中,fm索引指ferragina-manzini索引,核心在于构建基于bwt轮转字符串数组的lf映射,从bwt轮转字符串数组的某个位置映射到原始字符串的相应位置;
7、(4)计算参考序列的所有后缀,排序后缀,得到后缀数组suffixes;
8、(5)计算后缀数组中相邻后缀的最长公共前缀,得到最长公共前缀数组lcp;
9、(6)对最长公共前缀数组进行采样,得到采样后的最长公共前缀数组slcp;
10、(7)对fm索引和采样的最长公共前缀数组slcp进行序列化,得到对应的二进制文件index.bwt和slcparray.bwt;
11、(8)从rna测序数据reads中加载指定数量数据,其中,每批次加载的数量为batchsize,当剩余数据量小于batchsize时,全部加载;
12、(9)初始化多线程threads[numthreads],设置每个线程的参数,包括线程需要处理的测序数据的开始下标start和结束下标end,其中,numthreads为查找最大精确匹配所使用的线程数量;
13、(10)创建多线程,每个线程查找所分配的测序数据与查询序列之间的最大精确匹配,其中,最大精确匹配指rna测序数据与参考序列之间最长的连续相等子串;
14、(11)同步多线程,等待所有线程执行结束;
15、(12)将每个线程查找到的最大精确匹配合并到输出查询结果文件里;
16、(13)重复步骤(8)至步骤(12),直至处理完数据集里所有的rna测序数据。
17、针对本发明涉及的并行查找算法,使用运行时间、加速比、内存峰值和每秒钟处理读段数作为评估指标。
18、本发明的有益效果是:提供了一种用于加速第三代rna测序数据比对的最大精确匹配并行查找算法。该算法提出了一种基于多线程的最大精确匹配并行查找策略,大大减少了在rna测序数据和基因组之间搜索最大精确匹配所需的时间,同时还对程序的内存使用进行了优化。此外,通过序列化用于查找最大精确匹配的参考基因组索引,几乎消除了索引创建的时间开销,因此能够有效地处理大规模数据集。将该最大精确匹配并行查找算法应用在rna测序数据剪接比对算法的种子步骤中,在不影响算法整体准确率的情况下,显著减少程序运行时间。
技术特征:
1.一种用于加速第三代rna测序数据比对的最大精确匹配并行查找算法,其特征在于,包括以下步骤:
技术总结
本发明公开了一种用于加速第三代RNA测序数据比对的最大精确匹配并行查找算法,适用于对以“索引‑种子‑链接”为设计思想的比对算法进行加速。该算法首先对基因组的参考序列进行BWT变换,构建对应的FM索引;然后计算基因组参考序列的所有前缀,并对它们进行排序,从而创建最长公共前缀数组并采样。为了能加快查找最大精确匹配,采用基于POSIX的多线程进行并行化处理;此外,为了缩短程序的执行时间,对FM索引和最长公共前缀数组进行序列化。实验结果表明,该算法能极大加快最大精确匹配的查找,进而有效地加速RNA测序数据的剪接比对。
技术研发人员:王荣兴,张艳菊
受保护的技术使用者:桂林电子科技大学
技术研发日:
技术公布日:2024/3/11
- 还没有人留言评论。精彩留言会获得点赞!