一体化检测甲基化、CNV、单亲二体、三倍体和ROH的方法及装置与流程
本发明属于遗传检测,具体涉及一体化检测甲基化、cnv、单亲二体、三倍体和roh的方法及装置。
背景技术:
1、甲基化水平异常、拷贝数变异(cnv)、单亲二体(upd)、三倍体和存在连续性纯合片段(runs of homozygosity,roh)是导致许多常见遗传疾病、妊娠失败、癌症和其他复杂疾病的大型基因组畸变,同时识别这些畸变对于了解疾病至关重要。
2、拷贝数目变异也称拷贝数目多态,是一种大小介于1 kb至3 mb的dna片段的变异,在人类基因组中广泛分布,其覆盖的核苷酸总数大大超过单核苷酸多态性的总数,极大地丰富了基因组遗传变异的多样性。研究表明,孕妇群体中胎儿携带致病性拷贝数变异的比例可达1.6%~1.7%,远高于21、18、13-三体综合征0.2%的发生率。因此,对致病性cnvs的检测应作为产前诊断的重要内容之一。
3、正常人类细胞中包含两组染色体,一组来自父亲,一组来自母亲,三倍体是胎儿细胞中多了一组额外的染色体组,单亲二体是当一对的两条染色体都来自父亲或母亲一方时即为单亲二体,三倍体和单亲二体均是严重的染色体异常,是妊娠早期流产的重要原因之一。roh是一种基因组区域中一定范围内连续呈现的杂合性丢失的现象,染色体存在roh时提示可能存在upd,upd出现在特定的染色体上时,会由于遗传印记效应引起相关疾病。此外,roh区域内发生孟德尔隐性遗传病的风险明显增加。
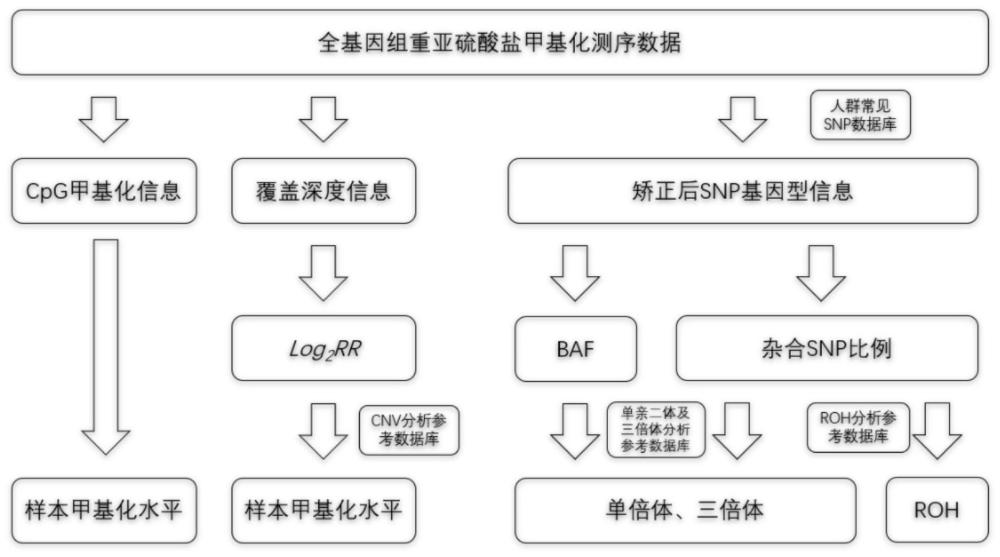
4、胚胎发育过程中的甲基化水平异常与妊娠失败具有一定的相关性,目前全基因组亚硫酸氢盐测序(wgbs)为甲基化检测的常规方法。该方法是在传统全基因组测序(wgs)的基础上使用亚硫酸氢盐将未甲基化的c转化为t,从而实现对基因组的甲基化水平进行检测。而wgs可以对拷贝数变异(cnv)、单亲二体(upd)、三倍体和存在连续性纯合片段(runsof homozygosity,roh)进行一体化地高效检测,但是无法对甲基化水平进行检测。
5、综上所述,现有技术中无有效方法进行低成本、高效地一体化检测,因此,开发一种对甲基化水平检测的同时对cnv、单亲二体、三倍体和roh进行一体化有效检测的方法,对于遗传检查领域具有重要意义。
技术实现思路
1、针对现有技术存在的不足,本发明的目的在于提供一体化检测甲基化、cnv、单亲二体、三倍体和roh的方法及装置。本发明基于全基因组重亚硫酸盐甲基化测序的wgbs的数据,在检测甲基化水平的同时也能够对cnv、单亲二体、三倍体和roh进行同步检测,实现一体化检测出甲基化、cnv、单亲二体、三倍体和roh区域,检测过程简单、成本低,对于遗传检测技术领域具有重要意义。
2、为达到此发明目的,本发明采用以下技术方案:
3、第一方面,本发明提供一体化检测甲基化、cnv、单亲二体、三倍体和roh的方法,所述方法包括:
4、(1)将样本进行全基因组甲基化测序,获取样本在基因组上cpg的甲基化信息和覆盖深度信息;从wgbs测序数据中获取未校正的snp(单核苷酸多态性)基因型信息,并构建校正模型对snp基因型信息进行校正,获取校正后的snp基因型信息;
5、(2)构建单亲二体及三倍体分析参考数据库、cnv分析参考数据库和roh分析参考数据库;进行甲基化水平分析、单亲二体及三倍体分析、cnv分析和roh分析。
6、本发明创造性地设计遗传检测分析流程,提出了一体化检测甲基化水平、cnv、单亲二体、三倍体和roh的方法,流程图如图1所示,所述方法能够基于全基因组重亚硫酸盐甲基化测序数据,一体化检测出甲基化水平、cnv、单亲二体、三倍体和roh区域,简化了检测流程,提高了检测效率且降低了检测成本。
7、本发明中,以获取的wgbs测序数据进行甲基化水平分析,以校正后的snp基因型信息进行cnv、单亲二体、三倍体和roh区域分析。所述校正后的snp基因型信息的获取如图2所示。
8、优选地,步骤(1)中,所述snp基因型信息包括snp位点的基因型和b等位基因频率(baf)。
9、优选地,步骤(1)中,所述获取样本在基因组上覆盖深度信息包括:将基因组分成每1 kb~100 kb一个的窗口,统计每个窗口的覆盖深度信息。
10、本发明中,所述覆盖深度信息包括样本基因组中窗口的覆盖深度,所述窗口的长度为1 kb~100 kb,包括但不限于2 kb、3 kb、4 kb、5 kb、6 kb、7 kb、8 kb、9 kb、10 kb、20kb、30 kb、40 kb、50 kb、60 kb、70 kb、80 kb、90 kb、91 kb、92 kb、93 kb、94 kb、95 kb、96kb、97 kb、98 kb或99 kb等。
11、优选地,步骤(1)中,所述校正模型包括常规校正模型和特殊校正模型。
12、优选地,所述常规校正模型的构建方法包括:通过隐马尔可夫模型(hiddenmarkov model,hmm),对不位于筛选后的人群常见snp数据库的snp基因型信息进行常规校正模型训练,从而获得常规校正模型。
13、优选地,所述特殊校正模型的构建方法包括:对位于筛选后的人群常见snp数据库的snp基因型信息进行以snp数据库为参考的特殊校正模型训练,从而获取特殊校正模型。
14、优选地,所述对未校正的snp基因型信息进行校正的步骤包括:对待测样本的读段比对后生成的bam文件进行校正:针对bam文件中不位于筛选后的人群常见snp数据库的读段,运用构建好的常规校正模型进行校正,从而消除因重亚硫酸盐而错误引入的snp;针对位于筛选后的人群常见snp数据库中的读段,运用构建好的特殊校正模型进行校正,从而校正受到重亚硫酸盐影响的snp的突变频率。
15、优选地,所述snp矫正模型的包括以下步骤:
16、(1’)获取人群常见snp数据库信息,并剔除其中无法矫正的snp突变位点,包括c突变为t的位置、t突变为c的位置、g突变为a的位置、a突变为g的位置。
17、(2’)对读段比对后生成的bam文件进行矫正,针对bam文件中‘xg’信息为‘ct’的读段(表明该读段的甲基化转化类型是c转化为t)进行t->c矫正,针对bam文件中‘xg’信息为‘ga’的读段(表明该读段的甲基化转化类型是g转化为a)进行g->a矫正。针对位于筛选后的人群常见snp数据库信息中位置的读段进行特殊模型矫正,具体方法是,针对bam文件中‘xg’信息为‘ct’的且snp数据库信息中ref或alt为c的读段的相关位置进行t->c矫正,针对bam文件中‘xg’信息为‘ga’的且snp数据库信息中ref或alt为g的读段的相关位置进行a->g矫正。
18、(3’)使用矫正后的bam文件进行snp检测,获取矫正后的snp基因型信息。
19、优选地,步骤(2)中,所述甲基化水平分析包括:
20、统计样本中单个cpg位点上覆盖到的测序结果为c的读段数量和测序结果为t的读段的数量,并按式(1)计算样本的甲基化水平mc_level;
21、式(1);
22、其中,ci为单个cpg位点上覆盖到的测序结果为c的读段数量,ti为单个cpg位点上覆盖到的测序结果为t的读段数量。
23、优选地,所述甲基化水平异常的判断标准为:正常胚胎样本的甲基化水平阈值范围17%-37%,正常细胞系、羊水或流产组织样本的甲基化水平阈值范围为50%-70%。
24、优选地,步骤(2)中,所述单亲二体及三倍体分析包括:
25、统计样本染色体的b等位基因频率偏移量和杂合snp比例,并按式(2)计算染色体的z值,其中,r1chr为样本染色体的r值,为单亲二体及三倍体分析参考数据库中相应染色体r值的平均值,σchr为单亲二体及三倍体分析参考数据库中染色体r值的标准差;
26、式(2)。
27、优选地,步骤(2)中,所述单亲二体及三倍体分析参考数据库的构建方法包括以下步骤:
28、(i)选取样本的snp基因型信息;
29、(ii)统计样本染色体的杂合snp的b等位基因频率与二倍体杂合snp的b等位基因频率理论值的距离,取平均值作为该条染色体的b等位基因频率偏移量offsetchr;
30、(iii)统计杂合snp比例fracchr;
31、(iv)整合染色体的b等位基因频率偏移量和杂合snp比例,按式(5)计算r值;
32、式(5)。
33、优选地,本发明基于校正后的snp基因型信息,所述单亲二体及三倍体分析参考数据库按性别分为男性、女性两类,所述单亲二体及三倍体分析参考数据库的构建方法包括以下步骤:
34、(i’)样本选择:选取多个二倍体样本的snp基因型信息;
35、(ii’)baf偏移量统计:理论上,若样本只存在一套遗传物质,则其所有snp均为纯合状态,baf为0或1;若样本存在两套遗传物质,则snp存在杂合、纯合两种状态,杂合snp的baf为0.5,纯合snp的baf为0或1;若样本存在三套遗传物质,则snp存在杂合、纯合两种状态,杂合snp的baf为0.33或0.66,纯合snp的baf为0或1;
36、每个样本分别统计其每条染色体的杂合snp的baf与二倍体杂合snp的baf理论值(0.5)的距离,取平均值作为该条染色体的baf偏移量offsetchr,如式(6)所示;
37、式(6);
38、(iii’)杂合snp比例统计:理论上,若样本只存在一套遗传物质,则其所有snp均为纯合状态,杂合snp比例为0;若样本存在二套遗传物质,在不考虑自然界遗传因素的情况下,其杂合snp比例为1/3;若样本存在三套遗传物质,在不考虑自然界遗传因素的情况下,其杂合snp比例为1/2;每个样本分别统计其每条染色体上杂合snp占该条染色体总snp的比例fracchr,如式(7)所示,其中nhet为杂合snp的数目,nhom为纯合snp的数目;
39、式(7);
40、(iv’)参数整合:整合每条染色体的baf偏移量和杂合snp比例,按式(4)计算r值。
41、优选地,步骤(2)中,所述单亲二体及三倍体分析包括以下步骤:
42、(i)统计样本染色体的b等位基因频率偏移量和杂合snp比例,并进行参数整合;
43、(ii)并按式(2)计算样本染色体的z值,z≥3,则该条染色体为三体;z≤-3,则该条染色体为单体;计算样本所有染色体的z值的中位数zgenome,zgenome≥3,则该样本为三倍体;zgenome≤-3,则该样本为单亲二体。
44、优选地,所述单亲二体及三倍体分析包括以下步骤:
45、(i’)按照上述构建单亲二体及三倍体分析参考数据库的方法统计样本每条染色体的baf偏移量与杂合snp比例,并进行参数整合;
46、(ii’)z-score标准化:结合样本与相应性别单亲二体及三倍体分析参考数据库中所有样本,对每条染色体的r值分别进行z-score标准化,按式(2)计算出每条染色体的z值,衡量该条染色体是否异常;
47、(iii’)异常报告:z≥3,则该条染色体为三体;z≤-3,则该条染色体为单体;计算样本所有染色体的z值的中位数zgenome,zgenome≥3,则该样本为三倍体;zgenome≤-3,则该样本为单亲二体。
48、优选地,步骤(2)中,所述cnv分析包括:
49、计算样本与cnv分析参考数据库比对结果log2rr的平均值,并利用所述平均值按式(3)计算拷贝数cn;
50、式(3)。
51、优选地,步骤(2)中,所述cnv分析参考数据库的构建方法包括以下步骤:
52、(a)选取样本的覆盖深度信息;
53、(b)采用定基比率法对窗口进行标准化;
54、(c)过滤掉覆盖深度一直为0或sd大于sd阈值的窗口;
55、(d)采用loess算法,对样本进行gc校正;
56、(e)将样本按照对应窗口合并,取平均值;
57、(f)按照步长3~5、滑动1~3的策略将窗口合并。
58、优选地,本发明基于测序数据的覆盖深度信息,所述cnv分析参考数据库按性别分为男性、女性两类,所述cnv分析参考数据库的构建方法包括以下步骤:
59、(a’)样本选择:选取多个二倍体样本的覆盖深度信息;
60、(b’)标准化:为了消除测序数据量的差别,以每个样本的窗口覆盖深度的中位数为基数,采用定基比率法对每个窗口进行标准化;
61、(c’)确定过滤阈值:计算每个窗口在多个样本中的sd,确定整体窗口的sd阈值;
62、(d’)窗口过滤:整合所有样本,过滤掉覆盖深度一直为0或在多个样本中sd大于sd阈值的窗口;
63、(e’)gc校正:整个基因组中,不同区域gc含量不同,而不同的gc含量可能会影响实验过程中片段的扩增效率,为了消除gc含量带来的误差,采用loess(局部加权回归)算法,对每个样本分别进行gc校正;
64、(f’)样本合并:将所有样本按照对应窗口合并,取平均值;
65、(g’)窗口合并:选择合适的分析策略,按照步长3~5、滑动1~3的策略(例如步长4、滑动1等)将小窗口合并为大窗口,大窗口值等于其包含的小窗口之和。
66、优选地,步骤(2)中,所述cnv分析包括以下步骤:
67、(a)采用定基比率法对窗口进行标准化;
68、(b)过滤掉覆盖深度为0或sd大于sd阈值的窗口;
69、(c)采用loess算法,对样本进行gc校正;
70、(d)按照步长3~5、滑动1~3的策略(例如步长4、滑动1等)将窗口合并;
71、(e)按式(8)计算样本与cnv分析参考数据库比对结果log2rr;
72、式(8);
73、其中,r1为cnv分析参考数据库窗口值,r2为样本窗口值;
74、(f)查找染色体的cnv断点;
75、(g)针对所述cnv断点,采用游程检验算法合并染色体的片段;
76、(h)计算样本与cnv分析参考数据库比对结果log2rr的平均值,并利用所述平均值按式(3)计算拷贝数cn。
77、优选地,步骤(2)中,所述cnv分析包括以下步骤:
78、(a’)标准化:为了消除测序数据量的差别,以每个样本的窗口覆盖深度的中位数为基数,采用定基比率法对每个窗口进行标准化;
79、(b’)窗口过滤:过滤掉cnv分析参考数据库中过滤的窗口(覆盖深度一直为0的窗口或sd大于sd阈值的窗口);
80、(c’)gc校正:采用loess(局部加权回归)算法,对每个样本分别进行gc校正;
81、(d’)窗口合并:选择合适的分析策略,按照步长3~5、滑动1~3的策略(例如步长4、滑动1等)将小窗口合并为大窗口,大窗口值等于其包含的小窗口之和;
82、(e’)与cnv分析参考数据库比对:设相应性别的cnv分析参考数据库中某个窗口的值为r1,处理样本该窗口的值为r2,按式(8)计算与cnv分析参考数据库的比对结果log2rr;
83、(f’)断点识别:采用环状二元分割算法(circular binary segmentation,cbs)或隐马尔可夫模型(hidden markov model,hmm)等算法查找每条染色体的cnv断点,将染色体划分为多个片段;
84、(g’)小片段合并:针对cbs或hmm等算法查找的cnv断点,对相同染色体的相邻片段采用游程检验算法进行合并,若检验的p-value>1e-5,说明相邻区域无显著差异,则将两个区域进行合并,再与下一个片段进行检验,依次类推,最终每个区域的log2rr值为该区域包含的大窗口的log2rr值的平均值;
85、(h’)拷贝数计算:正常人类细胞为二倍体,则每条染色体有两个拷贝,按式(3)计算样本区域的拷贝数cn;
86、(i’)异常区域报告:报告该样本中cn大于重复阈值线的区域与cn小于缺失阈值线的区域。
87、优选地,步骤(2)中,所述roh分析包括:
88、按式(4)计算样本与roh分析参考数据库的比对结果log10ff,进行断点识别和片段合并;
89、式(4);
90、其中,f1为roh分析参考数据库窗口值,f2为样本窗口值。
91、优选地,步骤(2)中,所述roh分析参考数据库的构建方法包括以下步骤:
92、(①)选取样本的snp基因型信息;
93、(②)将基因组划分为大小为400~800 kb的等分窗口(例如可以是410 kb、420 kb、450 kb、460 kb、500 kb、600 kb、650 kb、700 kb、750 kb、780 kb或790 kb),统计窗口的杂合snp比例;
94、(③)采用定基比率法对窗口进行标准化;
95、(④)将样本按照对应窗口合并,取平均值。
96、优选地,本发明基于校正的snp基因型信息,所述roh分析参考数据库按性别分为男性、女性两类,所述roh分析参考数据库的构建方法包括以下步骤:
97、(①’)样本选择:选取多个二倍体样本的snp基因型信息;
98、(②’)杂合snp比例统计:将基因组划分为600 kb窗口,按式(7)统计每个窗口的杂合snp比例;
99、(③’)窗口标准化:以每个样本的窗口覆盖深度的中位数为基数,采用定基比率法对每个窗口进行标准化;
100、(④’)样本合并:将所有样本按照对应窗口合并,取平均值。
101、优选地,步骤(2)中,所述roh分析包括以下步骤:
102、(1’)按照roh分析参考数据库的构建方法统计窗口的杂合snp比例;
103、(2’)按式(4)计算样本与roh分析参考数据库的比对结果log10ff;
104、(3’)查找染色体的cnv断点;
105、(4’)针对所述cnv断点,采用游程检验算法合并染色体的片段,报告样本中log10ff小于阈值线的区域。
106、优选地,步骤(2)中,所述roh分析包括以下步骤:
107、(1’’)按照roh分析参考数据库的构建方法统计每个窗口的杂合snp比例;
108、(2’’)与roh分析参考数据库比对:设相应性别的roh分析参考数据库中某个窗口的值为f1,样本该窗口的值为f2,按式(4)计算与roh分析参考数据库的比对结果log10ff;
109、(3’’)断点识别:采用环状二元分割算法(circular binary segmentation,cbs)或隐马尔可夫模型(hidden markov model,hmm)等算法查找每条染色体的cnv断点,将染色体划分为多个片段;
110、(4’’)小片段合并:针对cbs或hmm等算法查找的cnv断点,对相同染色体的相邻片段采用游程检验算法进行合并,若检验的p-value>1e-5,说明相邻区域无显著差异,则将两个区域进行合并,再与下一个片段进行检验,依次类推,最终每个区域的log10ff值为该区域包含的大窗口的log10ff值的平均值;
111、(5’’)异常区域报告:报告该样本中log10ff小于阈值线的区域。
112、本发明可对任意核酸样本进行分析检测,可应用于人体核酸样本分析检测,也可用于非疾病诊断和/或治疗为目的的体外基因组相关科学研究,如三倍体发生机制的研究、女性年龄与流产组织拷贝数变异的相关性研究等。
113、第二方面,本发明提供一体化检测甲基化、cnv、单亲二体、三倍体和roh的装置,所述装置包括:
114、(1)信息采集模块:将样本进行全基因组甲基化测序,获取样本在基因组上cpg的甲基化信息和覆盖深度信息;从wgbs测序数据中获取未校正的snp基因型信息,并构建校正模型对snp基因型信息进行校正;
115、(2)构建参考数据库和分析模块:构建单亲二体及三倍体分析参考数据库、cnv分析参考数据库和roh分析参考数据库;进行甲基化水平分析、单亲二体及三倍体分析、cnv分析和roh分析。
116、优选地,所述信息采集模块中,所述snp基因型信息包括snp位点的基因型和b等位基因频率。
117、优选地,所述信息采集模块中,所述获取样本在基因组上覆盖深度信息包括:将基因组分成每1 kb~100 kb一个的窗口,统计每个窗口的覆盖深度信息。
118、优选地,所述信息采集模块中,所述校正模型包括常规校正模型和特殊校正模型。
119、优选地,所述常规校正模型的构建方法包括:通过隐马尔可夫模型,对不位于筛选后的人群常见snp数据库的snp基因型信息进行常规校正模型训练,从而获得常规校正模型。
120、优选地,所述特殊校正模型的构建方法包括:对位于筛选后的人群常见snp数据库的snp基因型信息进行以snp数据库为参考的特殊校正模型训练,从而获取特殊校正模型。
121、优选地,所述对未校正的snp基因型信息进行校正的步骤包括:对待测样本的读段比对后生成的bam文件进行校正:针对bam文件中不位于筛选后的人群常见snp数据库的读段,运用构建好的常规校正模型进行校正,从而消除因重亚硫酸盐而错误引入的snp;针对位于筛选后的人群常见snp数据库中的读段,运用构建好的特殊校正模型进行校正,从而校正受到重亚硫酸盐影响的snp的突变频率。
122、优选地,所述构建参考数据库和分析模块中,所述甲基化水平分析包括:
123、统计样本中单个cpg位点上覆盖到的测序结果为c的读段数量和测序结果为t的读段的数量,并按式(1)计算样本的甲基化水平mc_level;
124、式(1);
125、其中,ci为单个cpg位点上覆盖到的测序结果为c的读段数量,ti为单个cpg位点上覆盖到的测序结果为t的读段数量。
126、优选地,所述甲基化水平异常的判断标准为:正常胚胎样本的甲基化水平阈值范围17%-37%,正常细胞系、羊水或流产组织样本的甲基化水平阈值范围为50%-70%。
127、优选地,所述构建参考数据库和分析模块中,所述单亲二体及三倍体分析包括:
128、统计样本染色体的b等位基因频率偏移量和杂合snp比例,并按式(2)计算染色体的z值,其中,r1chr为样本染色体的r值,为单亲二体及三倍体分析参考数据库中相应染色体r值的平均值,σchr为单亲二体及三倍体分析参考数据库中染色体r值的标准差;
129、式(2)。
130、优选地,所述构建参考数据库和分析模块中,所述cnv分析包括:
131、计算样本与cnv分析参考数据库比对结果log2rr的平均值,并利用所述平均值按式(3)计算拷贝数cn;
132、式(3)。
133、优选地,所述构建参考数据库和分析模块中,所述roh分析包括:
134、按式(4)计算样本与roh分析参考数据库的比对结果log10ff,进行断点识别和片段合并;
135、式(4);
136、其中,f1为roh分析参考数据库窗口值,f2为样本窗口值。
137、第三方面,本发明提供一种计算机设备,包括存储器和处理器,所述存储器存储有计算机程序,所述计算机程序执行第一方面所述的一体化检测甲基化、cnv、单亲二体、三倍体和roh的方法中的步骤。
138、第四方面,本发明提供一种计算机可读存储介质,其上存储有计算机程序,所述计算机程序执行第一方面所述的一体化检测甲基化、cnv、单亲二体、三倍体和roh的方法中的步骤。
139、本发明所述一体化检测甲基化水平、cnv、单亲二体、三倍体和roh检测装置、计算机设备和计算机可读存储介质,通过利用全基因组比对结果对染色体进行检测分析,获取基因组的甲基化水平的同时,利用校正snp算法消除重亚硫酸盐甲基化测序对于snp检测的影响,从而达到同时检测cnv、单亲二体、三倍体和roh的目的,不再使用额外实验手段。可以提高染色体的检测效率、降低检测成本,基于机器学习模型和相应分析算法,可以提高检测的覆盖率和准确性。
140、本发明所述的数值范围不仅包括上述列举的点值,还包括没有列举出的上述数值范围之间的任意的点值,限于篇幅及出于简明的考虑,本发明不再穷尽列举所述范围包括的具体点值。
141、相对于现有技术,本发明具有以下有益效果:
142、(1)本发明的一体化检测甲基化水平、cnv、单亲二体、三倍体和roh的方法,能够基于全基因组重亚硫酸盐甲基化测序数据,在检测基因组的甲基化水平的同时,利用校正snp算法消除重亚硫酸盐甲基化测序对于snp检测的影响,从而达到同时检测cnv、单亲二体、三倍体和roh区域的目的,简化了检测流程,提高了检测效率且降低了检测成本。
143、(2)本发明所述一体化检测甲基化水平、cnv、单亲二体、三倍体和roh检测装置、计算机设备和计算机可读存储介质,通过利用全基因组比对结果对染色体进行检测分析,不使用额外实验手段。可以提高染色体的检测效率、降低检测成本,基于机器学习模型和相应分析算法,可以提高检测的覆盖率和准确性。
- 还没有人留言评论。精彩留言会获得点赞!