核酸适配体-多价药物偶联物及其制备方法与应用
本发明涉及医药化工领域,具体涉及核酸适配体-多价药物偶联物及其制备方法和应用。
背景技术:
1、癌症严重威胁着人类的健康和生命,已成为死亡的主要原因。癌症治疗主要包括传统的手术治疗、放射治疗、药物治疗和近年来发展迅速的靶向治疗、免疫治疗等新型治疗方式。临床上应用的抗肿瘤药物种类繁多,但随着美国国家癌症研究所(nci)植物筛选计划的展开和我国得天独厚的中医药背景,天然抗肿瘤药物已成为国内外研究的热点,先后共有13类天然小分子药物及其衍生物在不同国家获批上市,而且至今为临床上常用的化疗药物。天然抗肿瘤药物作为常用的药物形式,却由于其水溶性差,选择性差,生物利用度低等问题造成的严重毒副作用,如喜树碱、紫杉醇、水仙碱、鬼臼毒素等。
2、核酸适配体是一类具有特定三维构象的高水溶性rna或单链dna,其长度一般为20-80个碱基,可特异性结合目标蛋白、多肽等生物大分子,主要包括:dna、rna、修饰的dna以及修饰的rna。适配体较抗体具有水溶性好;结构稳定;筛选过程简单,周期短;相对分子质量小,具有快速的血浆清除率、强大的组织穿透力、免疫原性低、变性可逆等;易于合成修饰,实现多层面调控等优点。核酸适配体可与药物直接结合形成靶向药物传递体系,也可作为靶向基团修饰到药物载体表面,从而实现药物的靶向传递,为提高药物的临床疗效,降低毒副作用提供了一种可行的方法。
3、然而,现有技术中的核酸适配体-药物偶联物均为单价药物配体与受体结合的模式,而细胞表面受体蛋白总数是有限的,故目前的核酸适配体-药物偶联物效果仍然有限。
技术实现思路
1、针对以上技术现状,本发明的目的在于克服现有技术中所存在的适配体偶联物中载药量低,在体内肿瘤环境下释放量不足,进而造成血药浓度低导致的肿瘤抑制效果差的弊端,提供一种偶联多价药物的适配体偶联物。通过将小分子毒性药物与能够负载多分子药物的连接键连接,一方面将核酸适配体与多价药物偶联形成自组装聚合物前药,一方面达到多重载药的目的,例如载2分子、3分子、4分子甚至更多药物,从而实现小分子毒性药物抗肿瘤活性成倍的增加。另一方面,考虑到肿瘤微环境的特征主要包括整体缺氧、ph值低、高gsh、存在过表达的酶等,进一步在设计负载药物连接键时引入肿瘤微环境敏感基团,形成肿瘤微环境响应型连接键,以实现药物在肿瘤组织的智能释放。在上述构思基础上,确保偶联物在肿瘤部位释放原药,极大程度的降低药物毒性,并最大程度发挥高效低毒的抗肿瘤作用。为实现上述发明目的,本发明提供以下技术方案:
2、作为本发明的第一方面,本发明提供一种核酸适配体-多价药物偶联物,所述核酸适配体-多价药物偶联物具有如式(i)所示的结构:
3、a-r-b-c (i)
4、式(i)中,r为多价连接子,具有如式(ii)或式(iii)所示的结构:
5、(ii)
6、(iii)
7、其中,x为各自独立的n或c;
8、a为核酸适配体;
9、c为小分子毒性药物;
10、b为以下结构中的任意一种:-co-(ch2)n1-s-s-(ch2)n2-co-、-co-(ch2)n1-s-(ch2)n2-co-、-co-(ch2)n1-s-s-s-(ch2)n2-co-、-co-(ch2)n1-(ch2)n3-(ch2)n2-co-、-co-(ch2)n1-(o-ch2-ch2-o)n4-(ch2)n2-co-、-co-(ch2)n1-hn-nh-(ch2)n2-co-、-co-(ch2)n1-c2h2-co-(ch2)n2-co-。
11、其中n1、n2、n3、n4皆为整数,且1≤n1≤10,1≤n2≤10,1≤n3≤20,1≤n4≤20;优选2≤n1≤4,2≤n2≤4,2≤n3≤10,2≤n4≤10。
12、前述的核酸适配体-多价药物偶联物,所述r与一个或多个b通过酰胺键连接,当b为多个时,每个b相同或不同。所述b与c通过酯键相连接,当b为多个时,每个c相同或不同。
13、前述的核酸适配体-多价药物偶联物,所述式(ii)或式(iii)中,x所在的环为环己烷、苯、单氮杂苯、二氮杂苯、三氮杂苯中的一种;优选地,x所在的环为邻二氮杂苯、间二氮杂苯、对二氮杂苯、间三氮杂苯中的一种;更优选为哌啶、吡啶、嘧啶、哌嗪、吡嗪、哒嗪、1,3,5-间三氮杂苯中的一种。
14、前述的核酸适配体-多价药物偶联物,所述式(iii)中三个含有x的六元环相同或不相同。
15、前述的核酸适配体-多价药物偶联物,所述r为如下结构中的任意一种:
16、、、
17、、、
18、、
19、
20、
21、
22、。
23、前述的核酸适配体-多价药物偶联物,所述c为如下结构的中的一种或多种:
24、、
25、、、
26、;
27、其中,r1-r7分别独立地选自h、oh、c1-c6烷基、取代的c1-c6烷基、酰基、c1-c6烷氧基、c3-c6环烷基、取代的c3-c6环烷基、c3-c6杂环、c1-c6烷基巯基、c1-c6烷胺基或。
28、前述的核酸适配体-多价药物偶联物,所述c为如下结构中的一种或多种:
29、、、、
30、、、、
31、、、
32、、、、
33、、。
34、前述的核酸适配体-多价药物偶联物,所述a为如下种类的核酸适配体中的任意一种:
35、as1411、pegaptanib、sgc8c、a10、dnaaptamer、rnaaptamer、cl4、apt、e07,或上述核酸适配体的硫代磷酸酯或甲基酸酯,或上述核酸适配体中核糖上的2'与4'碳连结在一起形成的锁核酸。
36、前述适配体对应序列如下:
37、as1411:ggtggtggtggttgtggtggtggtgg
38、pegaptanib:gcgaaccgauggaauuuuuggacgcucgc
39、sgc8c:atctaactgctgcgccgccgggaaaatactgtacggttaga
40、a10:gggaggacgaugcggaucagccauguuuacgucacuccuugucaauccucaucggcagacgacucgcccga
41、dnaaptamer:gggagacaagaataaacgctcaa-(25n)-ttcgacaggaggctcacaacaggc
42、rnaaptamer:gggggcauacuugugagacuuuuaugucaccccc
43、cl4:gccuuaguaacgugcuuugaugucgauucgacaggaggc
44、apt:gcagttgatcctttggataccctgg
45、e07:ggacggauuuaaucgccguagaaaagcaugucaaagccggaaccgucc
46、作为优选,本发明中的化合物包含如下结构:
47、
48、
49、
50、
51、
52、为as1411、pegaptanib、sgc8c、a10、dnaaptamer、rnaaptamer、cl4、apt、e07中的任一种,优选为as1411。
53、作为本发明的第二方面,本发明提供所述核酸适配体-多价药物偶联物的制备方法。本发明所述核酸适配体-多价药物偶联物可以由本领域技术人员结合本发明记载的内容及本领域进行制备,本发明包括但不限于包含如下步骤(1)-(3)的制备方法:
54、(1)将式(i)中的小分子毒性药物c与b连接进行修饰;
55、(2)将经修饰的小分子毒性药物c与多重载药连接键r通过缩合剂连接,得到多价药物衍生物;
56、(3)将核酸适配体a溶解于水中,再将上述得到的多价药物衍生物与核酸适配体上的氨基进行反应,得到核酸适配体-多价药物偶联物。
57、优选地,上述方法进一步为如下步骤:
58、(1)将小分子毒性药物进行二硫键修饰;
59、(2)将经二硫键修饰的小分子毒性药物与多重载药的连接键通过缩合剂连接,得到多价药物衍生物;
60、(3)将核酸适配体溶解于水中,再将上述得到的多价药物衍生物用有机溶剂溶解,在缩合试剂作用下利用核酸适配体上的氨基进行反应得到核酸适配体-多价药物偶联物。
61、更优选地,所述(1)中的小分子毒性药物为具有前述核酸适配体-多价药物偶联物中c的结构,优选为喜树碱类似物或紫杉醇类似物。
62、更优选地,所述(2)中,以2,2'-二硫二吡啶为原料,先将2,2'-二硫二吡啶与3-巯基丙酸溶于有机溶剂(包括但不限于乙醇或甲醇),通过醋酸作用,使得巯基取代吡啶基团,重复两次,得到3,3'-二硫代二丙酸。再将小分子毒性药物(包括但不限于喜树碱或紫杉醇)溶于有机溶剂(包括但不限于二氯甲烷、dmf、dmso等)通过缩合剂(包括但不限于edci/dipea、dcc/dipea、dic/dipea等)与3,3'-二硫代二丙酸连接,得到二硫键修饰并带有羧基端的小分子毒性药物。具体结构如下:
63、、
64、更优选地,(1)为如下反应路线0:
65、
66、更优选地,上述反应路线0进一步为反应路线1或反应路线2:
67、反应路线1:
68、反应路线2:
69、
70、所述反应路线中的有机溶剂包括但不限于meoh、etoh、dmf、dmso、dcm、thf、acn、ea。
71、所述反应路线0、1、2中的化合物1、2的合成试剂用量包括但不限于0.1-10eq。
72、所述反应路线0、1、2中的反应温度为0-60 ℃,作为进一步优选方案,反应路线温度均为室温条件。
73、反应路线0、1、2中的反应结束时间包括但不限于2-48 h,例如2-36h、4-24h。作为进一步实施方案,反应路线0、1、2反应结束时间均以tlc和lc-ms检测为准(tlc和lc-ms检测反应至原料反应完全则证明反应结束)。
74、更优选地,步骤(2)中以3,5-二羟基苯甲酸甲酯为原料与n-boc溴乙胺在有机溶剂中(包括但不限于dmf或dmso)与碳酸钾反应,再通过碱性试剂(包括但不限于氢氧化锂或氢氧化钠)脱去甲酯,并与fmoc-乙二胺盐酸盐通过缩合剂反应(包括但不限于hatu/dipea或pybop/dipea),并通过盐酸/甲醇或三氟乙酸脱去boc基团,得到具有氨基端的载两分子药物的连接键,载四分子药物连接键步骤同上。具体结构如下:
75、载两分子药物连接键:
76、
77、载四分子药物连接键:
78、
79、进一步地,(2)可采用如下反应路线3、4:
80、反应路线3:
81、
82、反应路线4:
83、所述缩合试剂包括但不限于hatu/dipea、hatu/et3n、pybop/dipea、pybop/et3n、edci/dmap或eedq;
84、反应路线3、4中的有机溶剂包括但不限于dmf、dmso、dcm、thf、甲醇、乙醇、乙腈、叔丁醇、水或者它们任意的混合物。
85、反应路线3、4中的合成试剂的用量包括但不限于0.1-10eq。
86、反应路线3、4中的反应温度为0-80 ℃,作为进一步实施方案,优选50-80 ℃,优选50℃-80℃。
87、反应路线3、4中的反应结束时间包括但不限于2-48 h,例如2-36h、4-24h。作为进一步实施方案,反应路线3、4反应结束时间均以tlc和lc-ms检测为准(tlc和lc-ms检测反应至原料反应完全则证明反应结束)。
88、对于步骤(3),包括如下步骤:
89、a.将步骤(1)所得的经二硫键修饰的喜树碱类似物或紫杉醇类似物在有机溶剂中,与实施例2所得的能多重载药的连接键通过缩合剂连接,并在其他试剂作用下脱去fmoc保护基团,生成带有羧基端的多价药物;
90、b.将a所得多价药物溶于有机试剂,加入缩合剂,再将核酸适配体as141溶于去离子水中,并加入有机相中进行反应、即得。
91、进一步地,(3)可采用如下反应路线5、6、7、8:
92、反应路线5:
93、
94、反应路线6:
95、
96、反应路线7:
97、
98、反应路线8:
99、
100、反应路线5、6、7、8中喜树碱类似物和紫杉醇类似物的用量为10-200eq,作为进一步优选方案,优选用量为20eq或者50eq。
101、反应路线5、6、7、8中缩合剂的用量包括但不限于15-500eq,作为进一步优选方案,所述缩合剂的用量优选30eq。
102、反应路线5、6、7、8中反应温度包括但不限于10-60 ℃,作为进一步优选方案,所述温度优选37 ℃。
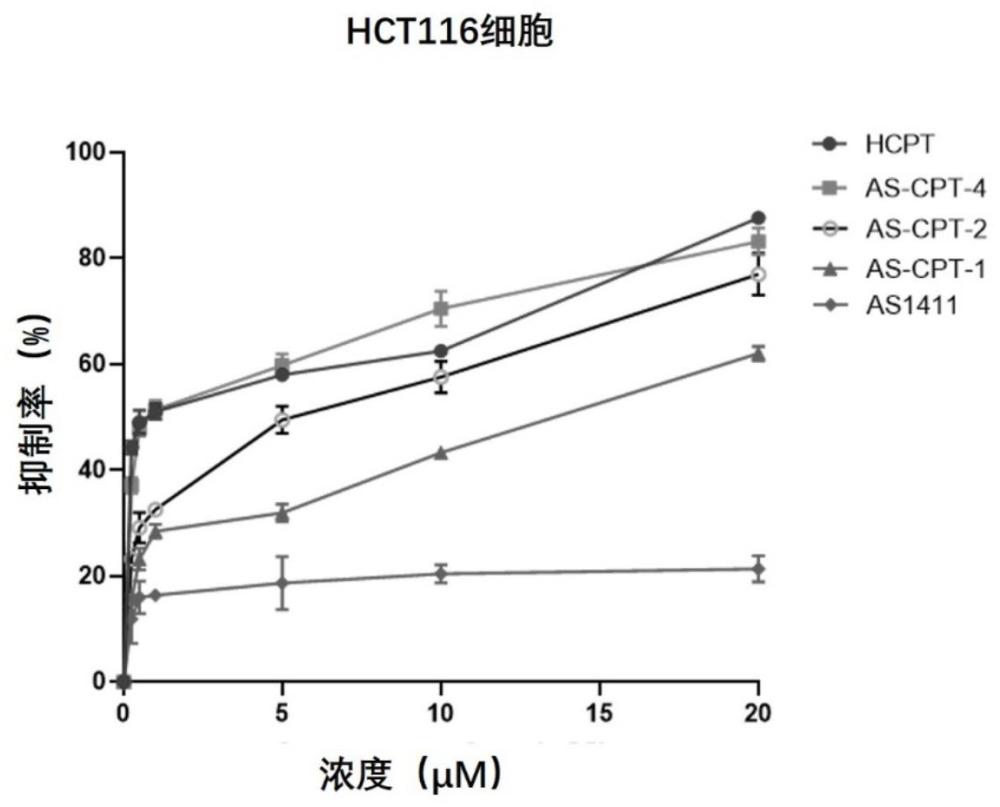
103、反应路线5、6、7、8中的反应结束时间包括但不限于2-48 h,例如2-36h、4-24h。作为进一步优选方案,反应路线5、6、7、8反应结束时间均以tlc和lc-ms检测为准(tlc和lc-ms检测反应至原料反应完全则证明反应结束,在与核酸适配体进行偶联时以hplc显示核酸适配体反应完全为准)。
104、本发明制备方法中所采用的各种分析、检测、及纯化方法均可以由本领域技术人员根据本发明公开内容并结合本领域常识进行确定,其并不影响本发明的实施。
105、作为本发明的第三方面,本发明还提供前述任一项核酸适配体-多价药物偶联物在制备抗肿瘤药物中的用途。优选地,所述肿瘤为结肠癌、乳腺癌、肝癌中的任意一种。
106、与现有技术相比,本发明的有益效果在于:
107、1.本发明提供的核酸适配体-多价药物偶联物中,核酸适配体-多价药物偶联物可自组装形成均一的纳米颗粒,提高小分子毒性药物的水溶性和生物相容性的同时,实现药物的主被动双靶向肿瘤细胞。
108、2.本发明提供的核酸适配体-多价药物偶联物中,实现单个核酸适配体载多个药物,达到多重载药的目的,大大提高药物的血药浓度,达到更好的肿瘤抑制效果。
109、3.本发明提供的核酸适配体-多价药物偶联物中,体外活性研究表明核酸适配体-多价药物偶联物抗肿瘤活性明显优于核酸适配体-单价药物偶联物,实现“1+1>4”的抗肿瘤模式。
110、4.本发明提供的核酸适配体-多价药物偶联物采用肿瘤微环境响应型连接键,在正常组织中无法释放小分子毒性药物;而在肿瘤组织中连接键断裂,释放小分子原药。相较于单纯的小分子毒性药物,偶合物对正常组织的毒性大大降低。
- 还没有人留言评论。精彩留言会获得点赞!