一种低共熔溶剂快速分离木质生物质主要组分的方法
1.本发明属于生物质精炼技术领域,具体涉及一种利用绿色低共熔溶剂一步处理实现木质生物质主要组分清洁分离的方法。
背景技术:
2.从绿色和可再生资源中清洁分离高附加值组分、生产清洁燃料和开发高性能替代材料一直以来被视为可持续经济的一部分,它对于解决能源短缺和环境污染问题具有重大意义。木质生物质作为地球上储量相当丰富的可再生资源,其被认为是化石能源的理想替代原料。木质生物质的主要组分包括纤维素、木质素和半纤维素,三组分之间错综复杂的联结形成了抗降解的“屏障”,为植物生长提供了力学支撑且能抵御微生物等的侵害,但这同时也为后续各组分分离及功能化应用带来了挑战。因此,高效打破木质生物质原料内部的“屏障”结构,快速分离其三组分,是实现生物质组分高值化应用的关键步骤。
3.传统的木质生物质组分分离技术包括酸化学法、碱化学法、热化学法、物理化学法和离子液体法等。然而这些方法存在以下缺点:强酸和强碱试剂的使用,对设备耐腐蚀要求高,同时反应过程中会产生大量废液,试剂回收困难且易造成环境污染,在高温等剧烈分离条件下,半纤维素和木质素结构易遭到破坏;离子液体等组分分离技术成本高,不利于工业化发展;大部分分离技术存在反应温度高,分离工艺复杂,能耗高等问题。
4.因此,针对上述传统分离技术存在的问题,开发新型的、绿色的、高效和低成本的分离技术是人们一直在研究的重要课题。
技术实现要素:
5.针对上述现有技术存在的问题,本发明的目的在于提供了一种用绿色低共熔溶剂一步法处理木质生物质原料快速分离主要组分的方法,所述分离方法环境友好、工艺简单、低能耗、成本低。所述分离方法包括以下步骤:
6.(1)将有机羧酸和多元醇按照1:1至1:8的摩尔比混合,40至80℃加热搅拌至形成均一透明的低共熔溶剂体系;
7.(2)将风干粉碎的生物质原料与步骤1)中制备的低共熔溶剂混合置于反应釜中,进行加热搅拌反应得到反应浆液,所述生物质原料与低共熔溶剂的固液重量比为1:5至1:30,反应温度为50至150℃,反应时间为1至36小时;
8.(3)反应结束后,冷却,加入乙醇/水的混合溶剂搅拌稀释,然后进行固液分离,所得固体干燥后得富纤维素组分;
9.(4)步骤(3)中经过固液分离后得到的滤液经旋转蒸发浓缩,加水沉淀、离心、洗涤、干燥获得木质素;
10.(5)步骤(4)中经过分离后得到的上清液再次经旋转蒸发浓缩,然后缓慢滴加到乙醇中沉淀、过滤、洗涤及干燥得到半纤维素。
11.(6)步骤(5)中分离半纤维素后的滤液经旋转蒸发除去乙醇和水分,即可回收低共
熔溶剂,回收的低共熔溶剂可循环利用。
12.优选地,步骤(1)中所述有机羧酸可选择草酸、二水合草酸、苹果酸、柠檬酸、马来酸、丙二酸、丁二酸的一种或多种。
13.优选地,步骤(1)中所述多元醇选用乙二醇、丙三醇、1,4-丁二醇、1,2-丙二醇的一种。
14.优选地,步骤(1)中所述有机羧酸和多元醇的摩尔比为1:3至1:6。
15.优选地,步骤(2)中所述风干粉碎的生物质原料选自针叶材、阔叶材或禾本科类植物原料,具体为10至80目的颗粒或长10至40mm且宽5至20mm的木片或竹片、或长10至50mm的禾本科原料等。
16.优选地,步骤(2)中生物质原料与低共熔溶剂的固液重量比为1:10至1:15。
17.优选地,步骤(2)中反应温度为70至120℃。
18.优选地,步骤(2)中反应时间为3至24小时。
19.优选地,步骤(3)中乙醇/水的混合溶剂中乙醇与水体积比为6:9至6:4。
20.优选地,步骤(3)中,基于步骤(2)的混合浆液的体积,所述乙醇/水的混合溶剂加入量为0.5至3倍体积。
21.优选地,步骤(4)和步骤(5)可不限制顺序,即分离完纤维素组分后,也可先实施步骤(5)分离半纤维素,再实施步骤(4)分离木质素。
22.有益效果
23.根据本发明的利用低共熔溶剂快速分离木质生物质主要组分的方法具有如下优点:
24.(1)所使用的低共熔溶剂由常见的有机酸和多元醇组成,其成本低廉、性质稳定、环境友好和可循环利用;
25.(2)仅需一步法处理,能有效破坏原料内部的“屏障”结构,同时实现纤维素、半纤维素、木质素三组分分离,该工艺简单,能耗低,实用性强,且过程中不使用有毒和不易回收的溶剂;
26.(3)该工艺所使用的低共熔溶剂酸性适中,且反应温度条件温和,避免了半纤维素的过度降解和木质素的缩合,可实现组分有效回收,获得纯度均大于90%的半纤维素和木质素。
附图说明
27.为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单的介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对本领域普通技术人员而言,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
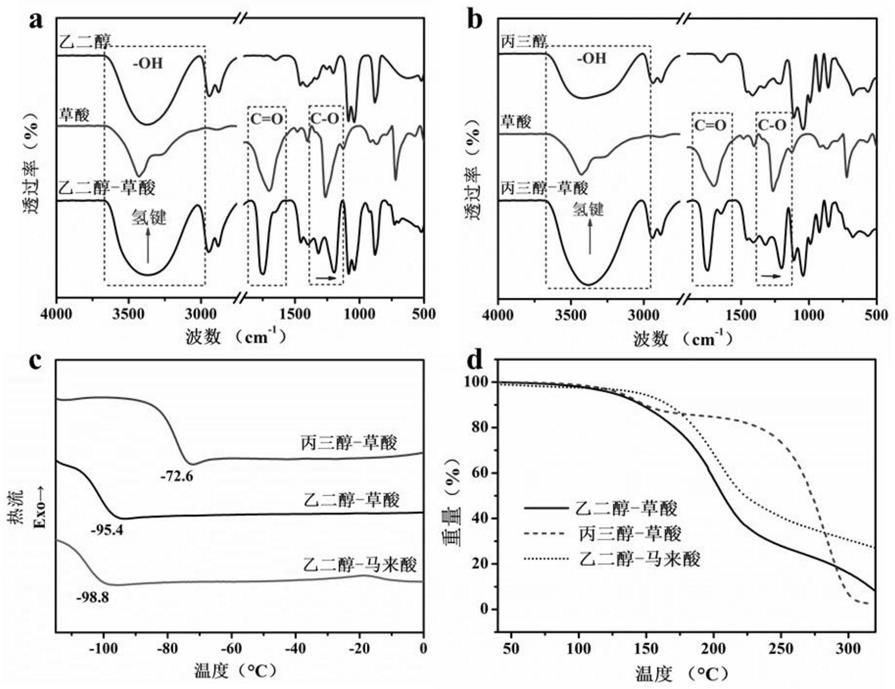
28.图1为实施例所用低共熔溶剂的部分结构及性质表征。其中(a)为实施例1~3和实施例6所用乙二醇-草酸低共熔溶剂及相应单组分化合物的ft-ir图;(b)为实施例4所用丙三醇-草酸低共熔溶剂及相应单组分化合物的ft-ir图;(c)为实施例1~6所用乙二醇-草酸、丙三醇-草酸和乙二醇-马来酸低共熔溶剂的dsc图;(d)为实施例1~5所用乙二醇-草酸、丙三醇-草酸和乙二醇-马来酸低共熔溶剂的tg图。
29.图2为实施例1、2、4、5分离得到的富纤维素(a)、半纤维素(b)和木质素(c)的ft-ir图。
30.图3为实施例2分离所得纤维素、半纤维素和木质素组分的照片。
具体实施方式
31.以下,将详细地描述本发明。在进行描述之前,应当理解的是,在本说明书和所附的权利要求书中使用的术语不应解释为限制于一般含义和字典含义,而应当在允许发明人适当定义术语以进行最佳解释的原则的基础上,根据与本发明的技术方面相应的含义和概念进行解释。因此,这里提出的描述仅仅是出于举例说明目的的优选实例,并非意图限制本发明的范围,从而应当理解的是,在不偏离本发明的精神和范围的情况下,可以由其获得其他等价方式或改进方式。
32.在本文中,所有以数值范围或百分比范围形式界定的特征或条件仅是为了简洁及方便。据此,数值范围或百分比范围的描述应视为已涵盖且具体公开所有可能的次级范围及范围内的个别数值,特别是整数数值。举例而言,“1至8”的范围描述应视为已经具体公开如1至7、2至8、2至6、3至6、4至8、3至8等等所有次级范围,特别是由所有整数数值所界定的次级范围,且应视为已经具体公开范围内如1、2、3、4、5、6、7、8等个别数值。除非另有指明,否则前述解释方法适用于本发明全文的所有内容,不论范围广泛与否。
33.若数量或其他数值或参数是以范围、较佳范围或一系列上限与下限表示,则其应理解成是本文已特定公开了由任一对该范围的上限或较佳值与该范围的下限或较佳值构成的所有范围,不论这些范围是否有分别公开。此外,本文中若提到数值的范围时,除非另有说明,否则该范围应包括其端点以及范围内的所有整数与分数。
34.在本文中,在可实现发明目的的前提下,数值应理解成具有该数值有效位数的精确度。举例来说,数字40.0则应理解成涵盖从39.50至40.49的范围。
35.在根据本发明所述的分离方法的步骤(1)中,选用有机酸和多元醇按1:1至1:8的摩尔比混合,更优选为1:4至1:6,并加热直至形成稳定均一的透明液体。多元醇与有机羧酸之间形成了氢键相互作用,最终形成的低共熔溶剂体系的熔点显著低于各组分的熔点。例如草酸常温下为固体,熔点为101~102℃,但与多元醇复配后,可以实现在本发明的反应温度下为液态。此外,通过调整低共熔溶剂各组分的配比来调控低共熔溶剂的性能,以实现从木质生物质原料中高效和选择性地分离各主要组分。
36.在根据本发明所述的分离方法的步骤(2)中木质生物质原料与低共熔溶剂的固液重量比为1:5至1:30,当重量比大于1:5时,反应体系粘度大,不利于传质,导致反应效果不理想;当重量比小于1:30时,大量的溶剂使用导致资源浪费,使成本增加,且单位溶剂效率低,不利于实际工业化。此外,反应温度选用70至120℃,较低的温度不利于原料中化学键的断裂,组分分离效率低。过高的温度一方面会破坏半纤维素的结构并生成低聚糖、单糖或小分子降解产物,难以从反应液中回收半纤维素,这会限制半纤维素组分的高值化利用。另一方面,在过高的温度及酸性条件下木质素会发生降解形成活性醛中间体,该中间体会进一步缩合形成顽固的c-c键,这极不利于木质素进一步转化为苯酚等酚类化合物的高值化利用。反应时间优选3至24小时。
37.本发明所述的分离方法的步骤(4)和步骤(5)可不限制顺序,即分离完纤维素组分
h伸缩振动,1734cm-1
处为富纤维素组分中残留的半纤维素上的乙酰基或糖醛酸中的c=o或酸性低共熔溶剂处理过程中纤维素羟基酯化后形成的酯的c=o振动,1603、1512和1428cm-1
处的低强度信号为木质素的特征吸收,表明其中含有少量木质素。在1174~1051cm-1
范围内的宽振动带主要来源于纤维素的c-o-c拉伸。
52.参照附图2中(b)图中a线,3418cm-1
为半纤维素中-oh的伸缩振动,2930cm-1
为c-h伸缩振动,1734cm-1
为半纤维素中乙酰基和糖醛酸中c=o的特征吸收峰,1640cm-1
为半纤维素吸附水的信号峰,1251cm-1
为葡萄糖醛酸中c-o振动的吸收峰,1046cm-1
为木聚糖的特征吸收峰,899cm-1
为c-1基团频率振动或环频率振动所产生,表明该低共熔溶剂分离得到的半纤维素糖基单元之间以β-糖苷键连接。
53.参照附图2中(c)图中a线,3412cm-1
为木质素中-oh的伸缩振动,2938cm-1
为c-h伸缩振动,1601、1512和1425cm-1
为木质素组分的芳环骨架振动,1459cm-1
为c-h变形振动和苯环振动,1329cm-1
处为紫丁香基苯环的c-o伸缩振动。1169cm-1
是酯中羰基的特征吸收谱带,是禾本科植物木质素中对羟苯基结构特有的吸收峰。1040cm-1
为愈创木基苯环吸收峰,834cm-1
处吸收峰由苯环上c-h振动产生。综上表明,该低共熔溶剂分离的木质素为典型的gsh型木质素。
54.实施例2
55.低共熔溶剂的配制:将乙二醇和草酸按照摩尔比1:4混合,置于密封容器中,60℃加热搅拌2小时,即得到均一透明的低共熔溶剂。
56.称取2g风干粉碎的玉米芯(40-60目)与上述制备的低共熔溶剂按1:15固液重量比混合加入反应釜,90℃下加热、搅拌反应6小时。反应结束后加入30ml重量百分百浓度为50%的乙醇溶液,充分混合后经离心分离,所得固体经数次乙醇溶液洗涤即得富纤维素组分。上清液经旋转蒸发浓缩至约40ml,然后缓慢滴加到3倍体积的乙醇中,静置沉淀,过滤、洗涤、干燥得到半纤维素。滤液经旋转蒸发除去乙醇,然后在搅拌下向其中加入250ml去离子水,沉淀过夜后经离心分离,所得固体经数次水洗后即得木质素。所得上清液经浓缩除去水后即可回收低共熔溶剂,回收的低共熔溶剂可再次利用。
57.经检测,富纤维素组分得率为46.5%,纯度为81.8%;半纤维素得率为24.5%,纯度为94.2%;木质素得率为53.6%,纯度为98.2%;低共熔溶剂回收率为96.9%。
58.参照附图2中(a)图中b线,3395cm-1
为富纤维素中-oh的伸缩振动,2897cm-1
为c-h伸缩振动,1734cm-1
处为富纤维素组分中残留的半纤维素上的乙酰基或糖醛酸中的c=o或酸性低共熔溶剂处理过程中纤维素羟基酯化后形成的酯的c=o振动,1603、1512和1428cm-1
处的低强度信号为木质素的特征吸收,表明其中含有少量木质素。在1174~1051cm-1
范围内的宽振动带主要来源于纤维素的c-o-c拉伸。
59.参照附图2中(b)图中b线,3418cm-1
为半纤维素中-oh的伸缩振动,2930cm-1
为c-h伸缩振动,1734cm-1
为半纤维素中乙酰基和糖醛酸中c=o的特征吸收峰,1640cm-1
为半纤维素吸附水的信号峰,1251cm-1
为葡萄糖醛酸中c-o振动的吸收峰,1046cm-1
为木聚糖的特征吸收峰,899cm-1
为c-1基团频率振动或环频率振动所产生,表明该低共熔溶剂分离得到的半纤维素糖基单元之间以β-糖苷键连接。
60.参照附图2中(c)图中b线,3412cm-1
为木质素中-oh的伸缩振动,2938cm-1
为c-h伸缩振动,1601、1512和1425cm-1
为木质素组分的芳环骨架振动,1459cm-1
为c-h变形振动和苯
环振动,1329cm-1
处为紫丁香基苯环的c-o伸缩振动。1169cm-1
是酯中羰基的特征吸收谱带,是禾本科植物木质素中对羟苯基结构特有的吸收峰。1032cm-1
为愈创木基苯环吸收峰,834cm-1
处吸收峰由苯环上c-h振动产生。综上表明,该低共熔溶剂分离的木质素为典型的gsh型木质素。
61.参考附图3,乙二醇-草酸在90℃下处理后实现纤维素、半纤维素和木质素三组分分离,由于反应条件温和,纤维素和半纤维素均显较白的颜色,且此条件下分离的木质素因没有发生缩合而显示较浅的颜色。
62.实施例3
63.低共熔溶剂的配制:将乙二醇和草酸按照摩尔比1:4混合,置于密封容器中,60℃加热搅拌2小时,即得到均一透明的低共熔溶剂。
64.称取2g风干粉碎的杨木粉(40-60目)与上述制备的低共熔溶剂按1:15固液重量比混合加入反应釜,110℃下加热、搅拌反应9小时。反应结束后加入30ml重量百分百浓度为50%的乙醇溶液,充分混合后经离心分离,所得固体经数次乙醇溶液洗涤即得富纤维素组分。上清液经旋转蒸发浓缩至约50ml,然后在搅拌下向其中加入250ml去离子水,沉淀过夜后经离心分离,所得固体经数次水洗后即得木质素。上清液经旋转蒸发浓缩至约40ml,然后缓慢滴加到3倍体积的乙醇中,静置沉淀,过滤、洗涤、干燥得到半纤维素。所得滤液经浓缩去除乙醇和水分后即可回收低共熔溶剂,回收的低共熔溶剂可再次利用。
65.经检测,富纤维素组分得率为50.5%,纯度为81.3%;半纤维素得率为28.6%,纯度为92.1%;木质素得率为55.0%,纯度为97.2%;低共熔溶剂回收率为92.7%。
66.实施例4
67.低共熔溶剂的配制:将丙三醇和草酸按照摩尔比1:4混合,置于密封容器中,60℃加热搅拌2小时,即得到均一透明的低共熔溶剂。
68.称取2g风干粉碎的玉米芯(40-60目)与上述制备的低共熔溶剂按1:15固液重量比混合加入反应釜,90℃下加热、搅拌反应12小时。反应结束后加入30ml重量百分百浓度为50%的乙醇溶液,充分混合后经离心分离,所得固体经数次乙醇溶液洗涤即得富纤维素组分。上清液经旋转蒸发浓缩至约50ml,然后在搅拌下向其中加入250ml去离子水,沉淀过夜后经离心分离,所得固体经数次水洗后即得木质素。上清液经旋转蒸发浓缩至约40ml,然后缓慢滴加到3倍体积的乙醇中,静置沉淀,过滤、洗涤、干燥得到半纤维素。所得滤液经浓缩去除乙醇和水分后即可回收低共熔溶剂,回收的低共熔溶剂可再次利用。
69.经检测,富纤维素组分得率为44.7%,纯度为81.9%;半纤维素得率为30.6%,纯度为94.4%;木质素得率为52.4%,纯度为97.6%;低共熔溶剂回收率为96.3%。
70.参照附图1中(b)图,本实施例所用丙三醇-草酸及单组分化合物的ft-ir谱图可知,纯丙三醇谱图中3373cm-1
为-oh的伸缩振动,而丙三醇-草酸谱图中该位置吸收谱带变宽,表明体系内形成了高度缔合的氢键。纯草酸谱图中1260cm-1
为c-o伸缩振动,而丙三醇-草酸谱图中该吸收峰发生红移,表明草酸结构上c-o中的o参与了与丙三醇之间的分子间相互作用(氢键)。参考附图1中(c)图中丙三醇-草酸的dsc曲线,形成的溶剂体系的熔点为-72.6℃,明显低于纯丙三醇(17.8℃)和纯草酸(101℃)的熔点,证明形成了低共熔溶剂。参考附图1中(d)图,丙三醇-草酸的分解温度在120℃以上,因此在该实施例中90℃下溶剂体系性质稳定。
71.参照附图2中(a)图中c线,3395cm-1
处为富纤维素中-oh的伸缩振动,2897cm-1
为c-h伸缩振动,1734cm-1
处为富纤维素组分中残留的半纤维素上的乙酰基或糖醛酸中的c=o或酸性低共熔溶剂处理过程中纤维素羟基酯化后形成的酯的c=o振动,1603、1512和1428cm-1
处的低强度信号为木质素的特征吸收,表明其中含有少量木质素。在1174~1051cm-1
范围内的宽振动带主要来源于纤维素的c-o-c拉伸。
72.参照附图2中(b)图中c线,3418cm-1
为半纤维素中-oh的伸缩振动,2930cm-1
为c-h伸缩振动,1734cm-1
为半纤维素中乙酰基和糖醛酸中c=o的特征吸收峰,1640cm-1
为半纤维素吸附水的信号峰,1251cm-1
为葡萄糖醛酸中c-o振动的吸收峰,1046cm-1
为木聚糖的特征吸收峰,899cm-1
为c-1基团频率振动或环频率振动所产生,表明该低共熔溶剂分离得到的半纤维素糖基单元之间以β-糖苷键连接。
73.参照附图2中(c)图中c线,3412cm-1
为木质素中-oh的伸缩振动,2938cm-1
为c-h伸缩振动,1601、1512和1425cm-1
为木质素组分的芳环骨架振动,1459cm-1
为c-h变形振动和苯环振动,1329cm-1
处为紫丁香基苯环的c-o伸缩振动。1169cm-1
是酯中羰基的特征吸收谱带,是禾本科植物木质素中对羟苯基结构特有的吸收峰。1040cm-1
为愈创木基苯环吸收峰,834cm-1
处吸收峰由苯环上c-h振动产生。综上表明,该低共熔溶剂分离的木质素为典型的gsh型木质素。
74.实施例5
75.低共熔溶剂的配制:将乙二醇和马来酸按照摩尔比1:4混合,置于密封容器中,60℃加热搅拌2小时,即得到均一透明的低共熔溶剂。
76.称取2g风干粉碎的玉米芯(40-60目)与上述制备的低共熔溶剂按1:20固液重量比混合加入反应釜,100℃下加热、搅拌反应6小时。反应结束后加入30ml重量百分百浓度为50%的乙醇溶液,充分混合后经离心分离,所得固体经数次乙醇溶液洗涤即得富纤维素组分。上清液经旋转蒸发浓缩至约50ml,然后在搅拌下向其中加入250ml去离子水,沉淀过夜后经离心分离,所得固体经数次水洗后即得木质素。上清液经旋转蒸发浓缩至约40ml,然后缓慢滴加到3倍体积的乙醇中,静置沉淀,过滤、洗涤、干燥得到半纤维素。所得滤液经浓缩去除乙醇和水分后即可回收低共熔溶剂,回收的低共熔溶剂可再次利用。
77.经检测,富纤维素组分得率为45.1%,纯度为83.4%;半纤维素得率为24.9%,纯度为94.0%;木质素得率为52.4%,纯度为98.7%;低共熔溶剂回收率为94.8%。
78.参考附图1中(c)图中乙二醇-马来酸的dsc曲线,形成的溶剂体系的熔点为-98.8℃,明显低于纯乙二醇(-12.9℃)和纯马来酸(135℃)的熔点,证明形成了低共熔溶剂。参考附图1中(d)图,乙二醇-马来酸的分解温度在140℃以上,因此在该实施例中100℃下溶剂体系性质稳定。
79.参照附图2中(a)图中d线,3395cm-1
处为富纤维素中-oh的伸缩振动,2897cm-1
为c-h伸缩振动,1734cm-1
处为富纤维素组分中残留半纤维素上的乙酰基或糖醛酸的c=o或酸性低共熔溶剂处理过程中纤维素羟基酯化后形成的酯的c=o振动,1603、1512和1428cm-1
处的低强度信号为木质素的特征吸收,表明其中含有少量木质素。在1174~1051cm-1
范围内的宽振动带主要来源于纤维素的c-o-c拉伸。
80.参照附图2中(b)图中d线,3418cm-1
为半纤维素中-oh的伸缩振动,2930cm-1
为c-h伸缩振动,1734cm-1
为半纤维素中乙酰基和糖醛酸中c=o的特征吸收峰,1640cm-1
为半纤维
素吸附水的信号峰,1251cm-1
为葡萄糖醛酸中c-o振动的吸收峰,1046cm-1
为木聚糖的特征吸收峰,899cm-1
为c-1基团频率振动或环频率振动所产生,表明该低共熔溶剂分离得到的半纤维素糖基单元之间以β-糖苷键连接。
81.参照附图2中(c)图中d线,3412cm-1
为木质素中-oh的伸缩振动,2938cm-1
为c-h伸缩振动,1601、1512和1425cm-1
为木质素组分的芳环骨架振动,1459cm-1
为c-h变形振动和苯环振动,1329cm-1
处为紫丁香基苯环的c-o伸缩振动。1169cm-1
是酯中羰基的特征吸收谱带,是禾本科植物木质素中对羟苯基结构特有的吸收峰。1031cm-1
为愈创木基苯环吸收峰,834cm-1
处吸收峰由苯环上c-h振动产生。综上表明,该低共熔溶剂分离的木质素为典型的gsh型木质素。
82.实施例6
83.低共熔溶剂的配制:将乙二醇和草酸按照摩尔比1:4混合,置于密封容器中,60℃加热搅拌2小时,即得到均一透明的低共熔溶剂。
84.称取2g风干粉碎的松木粉(40-60目)与上述制备的低共熔溶剂按1:20固液重量比混合加入反应釜,100℃下加热、搅拌反应24小时。反应结束后加入30ml重量百分百浓度为50%的乙醇溶液,充分混合后经离心分离,所得固体经数次乙醇溶液洗涤即得富纤维素组分。上清液经旋转蒸发浓缩至约50ml,然后在搅拌下向其中加入250ml去离子水,沉淀过夜后经离心分离,所得固体经数次水洗后即得木质素。上清液经旋转蒸发浓缩至约40ml,然后缓慢滴加到3倍体积的乙醇中,静置沉淀,过滤、洗涤、干燥得到半纤维素。所得滤液经浓缩去除乙醇和水分后即可回收低共熔溶剂,回收的低共熔溶剂可再次利用。
85.经检测,富纤维素组分得率为54.4%,纯度为77.8%;半纤维素得率为26.2%,纯度为90.5%;木质素得率为50.7%,纯度为96.5%;低共熔溶剂回收率为95.5%。
86.对比实施例1
87.称取2g风干粉碎的玉米芯(40-60目)与25%草酸溶液按1:15固液重量比混合加入反应釜,90℃下加热、搅拌反应12小时。反应结束后加入30ml重量百分百浓度为50%的乙醇溶液,充分混合后经离心分离,所得固体经数次乙醇溶液洗涤即得富纤维素组分。上清液经旋转蒸发浓缩至约40ml,然后缓慢滴加到3倍体积的乙醇中,静置沉淀,过滤、洗涤、干燥得到半纤维素。滤液经旋转蒸发除去乙醇,然后在搅拌下向其中加入400ml ph=2.0的盐酸水溶液,沉淀过夜后经离心分离,所得固体经数次水洗即得木质素。
88.经检测,富纤维素组分得率为71.5%,纯度为52.5%;半纤维素得率为5.2%,纯度为85.0%;木质素得率为26.6%,纯度为87.9%。可见草酸溶液处理效率低,处理后半纤维素和木质素组分得率及各组分纯度较实施例1~6低得多,且溶剂较低共熔溶剂更困难。
89.以上所述,仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应所述以权利要求的保护范围为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1