一种杂化钙钛矿量子阵列薄膜及其制备方法和用途
1.本发明涉及钙钛矿技术领域,尤其涉及一种杂化钙钛矿量子阵列薄膜及其制备方法和用途。
背景技术:
2.x射线检测广泛应用于医学诊断、工业无损检测、安全监控、核反应监测等领域。目前的x射线探测器有间接型x射线探测器与直接型x射线探测器两种。其中,直接型x射线探测器由半导体活性层和薄膜晶体管像素阵列组成,原理为半导体活性层将穿过被测物体的x射线直接转化为电信号,通过收集电信号获取被测物体信息,通过后处理成像。电荷的横向漂移和散射会导致收集的信号失真,限制其应用。
3.二维杂化钙钛矿的结构通式为a’a
n-1bn
x
3n+1
或a
’2a
n-1bn
x
3n+1
,其中,a’是大分子的有机配体,通常为有机铵的阳离子,a是甲胺、甲脒或铯等小尺寸阳离子,b是二价的金属离子,x是卤素阴离子,金属阳离子和卤素阴离子通过配位形成[bx6]八面体,a位阳离子分布在八面体共顶连接产生的空隙中,配位数为12,a’通过静电引力与八面体边缘的阴离子键合,形成有机间隔层。间隔层的介电常数与八面体片层失配,使[bx6]八面体层形成天然的量子阱,n表示量子阱的阱宽,即八面体层数。量子阱结构中载流子传输具有各向异性,通过对反应组分及制备工艺的调控能够控制二维杂化钙钛矿量子阱垂直基底生长,制备的薄膜中载流子沿着量子阱的生长方向定向迁移,同时能够抑制载流子的横向飘移和散射。这样独特的性质使得二维杂化钙钛矿在直接型x射线探测领域的高清成像领域具有巨大的应用价值。
[0004]
此外,不同阱宽的量子阱中激子结合能存在差异,影响电荷收集效率,因此除了保持垂直取向,获得具有均匀阱宽的量子阱是实现x射线探测器高清成像的关键。
[0005]
然而,目前对二维杂化钙钛矿垂直基底取向调控的研究多集中于太阳能电池,杂化钙钛矿薄膜的厚度为0.5μm左右,该厚度的薄膜对高能量的x射线吸收率低,产生的载流子较少,电信号弱,影响最终的成像清晰度。现有技术中,通过对有机配体、溶剂、添加剂的调控,控制二维杂化钙钛矿成核过程中八面体的聚集速度,同时调控成膜生长过程中溶剂及添加剂的挥发速度可以构建垂直于基底取向的量子阱阵列,然而这种常规方法易合成多n量子阱薄膜,不利于载流子均匀传输到阵列基底。非专利文献1中将乙酸与有机配体通过减压旋转蒸发合成氨基羧酸盐,接着将氨基羧酸盐与a位有机铵阳离子和b位的卤化物制备二维杂化钙钛矿的前驱体,采用浇铸的方法,将前驱体溶液旋涂于热基板上,制备垂直基底取向的量子阱。该方法中,乙酸能够与b位的阳离子形成强烈的配位作用,抑制[bx6]八面体的快速聚集,且在浇铸的过程中乙酸离子与过量的a位有机铵阳离子生成低蒸汽压的气体,随着溶剂匀速挥发,[bx6]八面体缓慢而均匀地聚集,形成单阱宽分布且垂直基底取向的薄膜,有望突破直接型x射线探测器高清成像的壁垒。然而,通过溶液热铸制备的膜层厚度有限,对x射线的吸收系数低,薄膜的成像效果差。将上述前驱体溶液通过刮涂技术提高薄膜的厚度至50μm,退火后得到的薄膜质量明显下降,出现了低阱宽且随机分布的量子阱,从而
降低x射线探测效率。
[0006]
非专利文献1:liang c,gu h,xia y,et al.two-dimensional ruddlesden-popper layered perovskite solar cells based on phase-pure thin films[j].nature energy,2021,6(1):38-45.
技术实现要素:
[0007]
针对上述技术问题,本发明提供一种应用于直接型x射线探测器的杂化钙钛矿量子阵列薄膜及其制备方法,本发明通过有机羧酸与有机胺制备的氨基羧酸盐,协同乙烯基酰胺类高分子添加剂制备杂化钙钛矿量子阵列薄膜,并将其用作直接型x射线探测器的量子阵列膜层。本发明提供的杂化钙钛矿量子阵列薄膜在保持二维杂化钙钛矿单阱宽分布和垂直基底取向的基础上通过对组分和制备工艺的调控增加了膜层厚度,基于其制备的直接型x射线探测器对x射线具有较高的吸收率,并且能够将载流子在量子阱中均匀传输到基底上,从而使被测物体的信号得到有效收集。
[0008]
为实现上述目的,本发明采取的技术方案为:
[0009]
一方面,本发明提供一种杂化钙钛矿量子阵列薄膜的制备方法,包括以下步骤:
[0010]
(1)将卤化铵盐(ax)、卤化铅(pbx2)、氨基羧酸盐和乙烯基酰胺类高分子于溶剂中反应,得到前驱体凝胶;
[0011]
(2)将步骤(1)得到的前驱体凝胶涂覆于基底上,加热除去溶剂后即得到所述杂化钙钛矿量子阵列薄膜。
[0012]
作为优选地实施方式,所述卤化铵盐(ax’)选自甲胺氢碘酸、甲脒氢碘酸、甲胺氢溴酸、甲脒氢溴酸、甲胺盐酸盐和甲脒盐酸盐中的任意一种或几种,优选为甲胺氢碘酸;
[0013]
优选地,所述卤化铅(pbx2)选自碘化铅、溴化铅和氯化铅中的任意一种或几种,优选为碘化铅;
[0014]
优选地,所述乙烯基酰胺类高分子选自聚乙烯吡咯烷酮、聚乙烯甲酰胺、聚乙烯己内酰胺和聚乙烯丙烯酰胺中的任意一种或几种,优选为聚乙烯吡咯烷酮。
[0015]
优选地,所述溶剂为二甲基亚砜和n,n-二甲基甲酰胺的混合溶剂;
[0016]
优选地,所述溶剂为二甲基亚砜和n,n-二甲基甲酰胺体积比为1:4~4:1的混合溶剂,进一步优选为二甲基亚砜和n,n-二甲基甲酰胺体积比为2:3或1:4的混合溶剂。
[0017]
作为优选地实施方式,所述氨基羧酸盐的制备方法包括将有机羧酸和有机胺在冰水浴中反应、减压旋转蒸发的步骤;
[0018]
优选地,所述有机羧酸选自乙酸、丙酸、正丁酸、异丁酸和正戊酸中的任意一种或几种,优选为丙酸和正丁酸中的任意一种或两种混合使用;
[0019]
优选地,所述有机胺选自正丁胺、异丁胺、α-苯乙胺、烯丙基胺、2-硫代-乙基胺中的任意一种或几种;
[0020]
优选地,所述有机羧酸和有机胺的摩尔比为1:2~2:1;
[0021]
优选地,所述在冰水浴中反应的时间为1~4h;
[0022]
优选地,所述减压旋转蒸发的温度为60~100℃,进一步优选为70~90℃;
[0023]
优选地,还包括重结晶的后处理操作。
[0024]
在某些具体的实施方式中,所述氨基羧酸盐的制备方法的具体操作为,将有机羧
酸和有机胺在冰水浴中搅拌2h,混合均匀,然后将混合物转移至减压旋转蒸发仪中,60~100℃,优选为70~90℃,旋转蒸发去除溶剂,收集蒸馏得到的产物;将得到的结晶产物使用乙醚洗涤后,用乙醇再次溶解,加入乙醚重结晶三次;得到的结晶产物再次溶于乙醇,在减压旋转蒸发仪中60~100℃,优选为70~90℃,旋转蒸发去除溶剂后,收集的液体产物即为高纯氨基羧酸盐,自然冷却备用。
[0025]
作为优选地实施方式,步骤(1)中,所述于溶剂中反应的方程式如式(i)所示:
[0026]
2a'oc+(n+1)ax'+npbx2——
→
a'2x'a
(n-1)
pbnx
(3n+1)
+2x'oc
↑(i)[0027]
式(i)中:a’oc为氨基羧酸盐,a’为有机铵根离子,oc为有机羧酸根离子;ax’为卤化铵盐,x’为卤离子;pbx2为卤化铅,x为卤离子;n=2~10,优选为4~10;
[0028]
优选地,所述卤化铵盐为甲胺氢碘酸,所述卤化铅为碘化铅,式(i)所示的反应方程式如式(ii)所示:
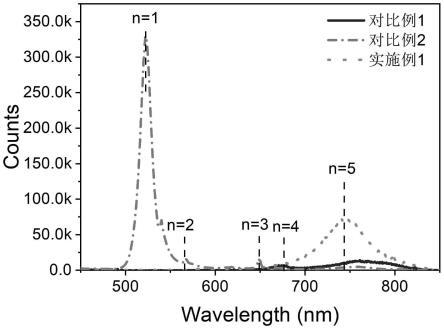
[0029]
2a’oc+(n+1)mai+npbi2——
→a’2ma
(n-1)
pb
ni(3n+1)
+2maoc
↑
(ii)
[0030]
式(ii)中:a’oc为氨基羧酸盐,a’为有机铵根离子,oc为有机羧酸根离子;mai为甲胺氢碘酸盐;pbi2为碘化铅;n=2~10,优选为4~10;
[0031]
优选地,所述碘化铅在反应体系中的浓度为0.5~4.5mol/l,优选为2.5~4mol/l;
[0032]
在本发明的技术方案中,所述乙烯基酰胺类高分子不参加反应,其分子中的羰基和oc中的羰基协同作用与pb
2+
形成中间体,该中间体的空间位阻较大,因此能够抑制[pbi6]八面体的快速聚集,降低生长速率,从而获得高结晶且取向的薄膜。因此,本发明中的乙烯基酰胺类高分子属于一种添加剂,即形成中间体的诱导分子,只有少量吸附在钙钛矿晶体表面,不作为反应物考虑;
[0033]
优选地,所述乙烯基酰胺类高分子的添加量为a'2x'a
(n-1)
pbnx
(3n+1)
总质量的1%~5%,例如为1%、2%、3%、4%、5%或它们之间的任意值,优选为1.5%~2%;
[0034]
优选地,步骤(1)中,所述于溶剂中反应为60~100℃搅拌反应1~4h;优选为70~80℃搅拌反应1~2h。
[0035]
作为优选地实施方式,步骤(2)中,所述涂覆为刮涂;
[0036]
优选地,所述刮涂的刮刀高度为50~300μm,进一步优选为150~200μm。
[0037]
在某些具体的实施方式中,所述基底需进行预处理,所述预处理为依次用去离子水、丙酮、乙醇和异丙醇进行超声洗涤后,用氮气吹干,在紫外臭氧清洗机中清洗0~20min,优选为10~15min。
[0038]
作为优选地实施方式,步骤(2)中,所述加热为程序升温;
[0039]
优选地,所述程序升温依次包括以下升温过程:
[0040]
(i)30℃加热2~10h,优选为8h;
[0041]
(ii)35℃加热2~10h,优选为8h;
[0042]
(iii)45℃加热2~12h,优选为10h;
[0043]
(iii)95℃加热0.5~2h,优选为1.5h;
[0044]
(iv)110℃加热2~8h,优选为6h。
[0045]
在本发明的技术方案中,程序升温能够控制溶剂的挥发速率,有利于形成具有载流子限域的量子阵列高质量薄膜。
[0046]
又一方面,本发明提供上述制备方法得到的杂化钙钛矿量子阵列薄膜。
[0047]
又一方面,本发明提供上述杂化钙钛矿量子阵列薄膜在制备直接型x射线探测器中的用途。
[0048]
又一方面,本发明提供一种直接型x射线探测器,包括基底、原位垂直生长于所述基底上的量子阵列膜层以及形成于所述量子阵列膜层上的电极层,其中,所述量子阵列膜层由上述杂化钙钛矿量子阵列薄膜构成,所述电极层形成于所述量子阵列膜层背离所述基底的面上。
[0049]
优选地,所述基底为氧化铟锡基底。
[0050]
优选地,所述电极层选自碳电极和金属电极中的至少一种,所述金属选自金、银和铜中的至少一种。
[0051]
优选地,所述电极层的制备方法包括如下步骤:
[0052]
将碳浆涂覆于所述量子阵列膜层上,固化;
[0053]
或,
[0054]
在所述量子阵列膜层上通过热蒸镀沉积金属。
[0055]
在某些具体的实施方式中,制备电极层时可以通过掩模版制备图案化电极层。
[0056]
优选地,所述固化为80~120℃下固化5~15min,进一步优选为90~110℃固化10~12min。
[0057]
上述技术方案具有如下优点或者有益效果:
[0058]
本发明提供的杂化钙钛矿量子阵列薄膜,以有机羧酸和有机胺合成的氨基羧酸盐,与卤化铵盐、卤化铅和乙烯基酰胺类高分子于溶剂中反应得到前驱体溶液,其中,乙烯基酰胺类高分子和羧酸中的羰基与pb
2+
八面体中心阳离子强烈的配位作用抑制了八面体的快速聚集,使[pbi6]匀速成核,同时有机羧酸的烷基链与高分子链之间的分子间作用力能够调控前驱体溶液的黏度,形成稳定的前驱体凝胶。接着将前驱体凝胶涂覆于在基底上,加热去除溶剂即可原位构筑厚度超过50μm以上单阱宽分布且垂直基底取向的薄膜,该过程中采用刮涂的方式时,二维杂化钙钛矿的八面体、层间有机阳离子通过分子间作用力自组装,垂直于基底生长,且基于羧酸与八面体中心阳离子之间的强烈的配位作用,使得热处理过程中溶剂和羧酸从体系中匀速挥发,在乙烯基酰胺类高分子添加剂的协同作用下,形成单阱宽分布的取向量子阱结构的杂化钙钛矿量子阵列薄膜,该量子阱结构有利于抑制x射线辐照下电信号的散射。在该薄膜表面制备电极层后得到的直接型x射线探测器,因量子阱的限域作用,载流子沿着量子阱均匀传输到像素基底上,有利于对待测物体的信息进行有效收集。
[0059]
与现有技术相比,本发明存在以下优点:
[0060]
(1)相对于现有技术仅适应500nm厚度的薄膜,提高薄膜厚度的话,会导致液膜溶剂在短时间内无法清除,薄膜质量下降的问题,本发明提供的杂化钙钛矿量子阵列薄膜,协同氨基羧酸盐和乙烯基酰胺类高分子与pb
2+
的相互作用,实现了量子阱薄膜垂直于基底生长、阱宽均匀分布窄,且厚度可达到50μm以上,同时能够明显改善薄膜质量。其中,氨基羧酸盐是保证量子阱薄膜垂直取向的关键,乙烯基酰胺类高分子中羰基的协同作用则是构建50μm以上的均匀阱宽薄膜的关键。其制备的直接型x射线探测器,对x射线的吸收强,降低x射线成像过程中载流子的横向散射,实现定向传输,高效收集电信号。
[0061]
(2)现有技术中,常规的热铸法中使用的高温退火使得液膜表层溶剂快速挥发,钙
钛矿量子阱在表面优先结晶,形成致密的固体膜,使溶剂挥发通道关闭,底层溶剂无法挥发从而导致薄膜质量急剧下降。而本发明使用的程序升温利用长时间的低温干燥以及后续的高温退火,控制微米级液膜中溶剂的挥发,能够在较长时间下保证溶剂挥发通道,且由于氨基羧酸盐和乙烯基酰胺类高分子的抑制结晶作用,溶剂均匀挥发,最终构建垂直且阱宽均匀的高质量微米级薄膜。
附图说明
[0062]
图1是实施例和对比例中制备的杂化钙钛矿量子阵列薄膜的荧光光谱图。
[0063]
图2是实施例和对比例中制备的杂化钙钛矿量子阵列薄膜的x射线衍射谱图。
[0064]
图3是实施例和对比例中制备的直接型x射线探测器在50kvp x射线开关下的i-t响应曲线。
具体实施方式
[0065]
下述实施例仅仅是本发明的一部分实施例,而不是全部的实施例。因此,以下提供的本发明实施例中的详细描述并非旨在限制要求保护的本发明的范围,而是仅仅表示本发明的选定实施例。基于本发明的实施例,本领域技术人员在没有作出创造性劳动的前提下所获得的所有其他实施例,都属于本发明的保护范围。
[0066]
在本发明中,若非特指,所有的设备和原料等均可从市场购得或是本行业常用的。下述实施例中的方法,如无特别说明,均为本领域的常规方法。
[0067]
实施例1
[0068]
本实施例中提供一种直接型x射线探测器,制备过程包括以下步骤:
[0069]
①
将摩尔比为1:1的乙酸与苯乙胺有机配体(a’)在0℃冰水浴中搅拌2h,混合均匀;然后将粗产物转移至减压旋转蒸发仪中,在60℃下旋转蒸发1h,去除溶剂,收集蒸馏得到的产物;接着将得到结晶产物使用乙醚洗涤三次,使用乙醇再次溶解、加入乙醚重结晶三次;得到的结晶产物再次溶于乙醇,在减压旋转蒸发仪中60℃下1h,收集得到的液体产物即为高纯苯乙胺羧酸盐,冷却至室温备用;
[0070]
②
将苯乙胺羧酸盐(peaac)、甲胺氢碘酸盐(mai)、碘化铅(pbi2)和聚乙烯吡咯烷酮溶解于二甲基亚砜和n,n-二甲基甲酰胺的混合溶剂中(二甲基亚砜和n,n-二甲基甲酰胺的体积比为1:4),70℃下加热搅拌2h,冷却得到前驱体凝胶;其中苯乙胺羧酸盐、甲胺氢碘酸盐(mai)、碘化铅(pbi2)的物质的量之比为2:6:5,聚乙烯吡咯烷酮的添加量为9.6mg,碘化铅在溶剂中的浓度为3.5mol/l,反应体系的总体积为0.2ml;
[0071]
③
将氧化铟锡基底先后使用去离子水、丙酮、乙醇和异丙醇在超声清洗器中洗涤,使用氮气流干燥后,在紫外臭氧清洗机中清洗15min;接着将前驱体凝胶滴加于氧化铟锡基底上,移动刮刀,刮刀高度为200μm,使前驱体凝胶在基底基板上形成均匀膜层;设置涂膜机程序温度为30℃加热8h,35℃加热8h,45℃加热11h,95℃加热1.5h,110℃加热2h,最终得到杂化钙钛矿量子阵列薄膜,其厚度为60μm。
[0072]
④
以杂化钙钛矿量子阵列薄膜作为量子阵列薄膜,并在其表面上使用热蒸镀的方法蒸镀铜电极,制备直接型x射线探测器。
[0073]
本实施例中制备的杂化钙钛矿量子阵列薄膜表现为单一的阱宽分布(图1),高结
晶度和垂直取向(图2),得到的x射线探测器具有最高的x射线光生电流(图3)。
[0074]
对比例1
[0075]
本对比例提供的直接型x射线探测器,制备过程同实施例1,区别在于省略步骤
①
,并将步骤
②
中的苯乙胺羧酸盐(peaac)替换成同物质的量的苯乙胺氢碘酸盐。
[0076]
本对比例中的杂化钙钛矿量子阵列薄膜的厚度为60μm。本实施例中制备的杂化钙钛pl强度较实施例低,且存在n=4和5的两种阱宽(图1),结晶度明显下降,且存在随机取向钙钛矿的衍射峰(图2)其作为x射线探测器具有较低的x射线光生电流,约为实施例1的一半(图3)。
[0077]
对比例2
[0078]
本对比例提供的直接型x射线探测器,制备过程同实施例1,区别在于步骤
②
中不添加聚乙烯吡咯烷酮。
[0079]
本对比例中的杂化钙钛矿量子阵列薄膜的厚度为60μm。与实施例1相比,本对比例因为缺少聚乙烯吡咯烷酮,得到的薄膜阱宽主要为n=1,同时存在n=2、3、4和5的弱pl峰,结晶度欠佳,由于低阱宽相的存在,其x射线衍射图谱中存在明显的随机取向二维杂化钙钛矿杂相,获得的x射线探测器具有明显的基线漂移,不利于对光生信号的高效收集。
[0080]
效果验证
[0081]
为了验证本发明的效果,对实施例以及对比例中制备的杂化钙钛矿量子阵列薄膜进行荧光光谱表征,激发波长为365nm,结果如图1所示:实施例1中量子阱阱宽集中分布在n=5处,符合原料的化学计量比。而对比例1中n=5的峰宽且强度低,阱宽在n=4处有明显分布,对比例2中量子阱阱宽主要分布于n=1处,同时存在n=2、3、4和5的量子阱。说明只有在peaac和聚乙烯吡咯烷酮协同作用下才能制备阱宽均匀分布的量子阱薄膜。
[0082]
为了验证本发明的效果,对实施例以及对比例中制备的杂化钙钛矿量子阵列薄膜进行x射线衍射谱图表征,结果如图2所示:实施例1中具有垂直取向二维杂化钙钛矿量子阱的三个明显的特征峰,分别表示晶面(111)、(202)和(313)。而对比例1中这三个特征峰的峰强明显降低,且存在随机取向的钙钛矿特征峰,在图中以“*”标注。对比例2中的峰强较实施例1略有下降,但较对比例1明显增强,同时存在随机取向的钙钛矿杂相。说明peaac是构建垂直取向钙钛矿量子阱的关键,其能够与聚乙烯吡咯烷酮协同作用,使低阱宽分布量子阱消失,从而得到结晶度高且垂直取向的量子阱薄膜。
[0083]
为验证peaac和聚乙烯吡咯烷酮的协同作用,将实施例和对比例制备的x射线探测器直接置于50kvp x射线开关和30v偏压下,测量i-t曲线,对比不同阱宽分布薄膜的x射线响应性,结果如图3所示:实施例1中peaac和聚乙烯吡咯烷酮协同制备的探测器x射线响应最强,且暗电流较低,且具有平稳的基线,说明peaac和聚乙烯吡咯烷酮协同作用能够提高薄膜质量,从而提高x射线的探测性能。
[0084]
以上所述仅是本发明的优选实施方式,应当指出:对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1