非复制型转导颗粒和基于转导颗粒的报告系统的制作方法
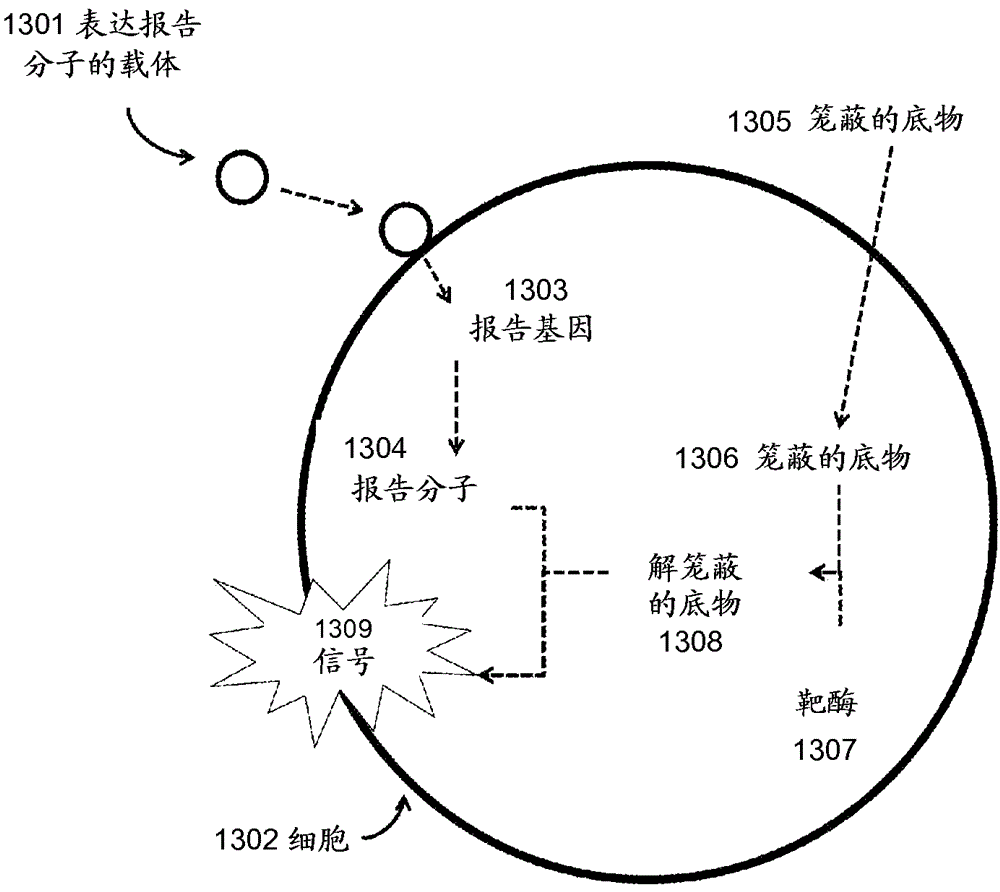
相关申请的交叉引用本申请要求于2013年3月13日提交的美国临时申请号61/779,177、2013年10月29日提交的美国临时申请号61/897,040,和2013年2月12日提交的美国临时申请号61/939,126的权益,其各自通过引用全部结合到本文中。发明背景。发明领域本发明涉及将非复制型转导报告分子包装和递送到细胞中以检测细胞中的靶基因的方法和组合物。相关技术的描述转导颗粒是指能够将非病毒核酸递送到细胞中的病毒。基于病毒的报告系统已经用于检测细胞的存在并且依赖于病毒的溶原化相以允许细胞表达报告分子。这些基于病毒的报告系统使用复制型(replication-competent)转导颗粒,其表达报告分子并导致靶细胞发出可检测信号。然而,病毒的裂解周期已经显示出对基于病毒的报告测定是有害的。carriere,c.等人,conditionallyreplicatingluciferasereporterphages:improvedsensitivityforrapiddetectionandassessmentofdrugsusceptibilityofmycobacteriumtuberculosis(条件复制型萤光素酶报告噬菌体:提高快速检测的灵敏性并评价结核分枝杆菌的药敏性).journalofclinicalmicrobiology,1997.35(12):第3232-3239页。carrière等人开发了结核分枝杆菌/卡介苗(bcg)萤光素酶报告噬菌体,其裂解周期在30°c受到抑制、但在37°c活化。使用该系统,carrière等人已经证明了使用具有被抑制的裂解周期的报告噬菌体来检测bcg。然而,在基于噬菌体的报告测定中抑制、而非消除噬菌体的复制功能,伴随不利之处。首先,控制噬菌体的复制功能会强加限制测定条件。例如,当噬菌体在30°c的非容许温度用于感染细胞时,carrière等人使用的报告噬菌体phae40的裂解周期是被阻遏的。该温度需求对报告测定强加限制条件,其中靶细菌的最佳温度为37°c。这些限制条件阻碍了最佳测定表现。此外,难以控制病毒的复制功能。在使用转导颗粒作为报告系统期间,病毒的复制应当被抑制。例如,carrière等人所报告的报告噬菌体phae40的裂解活性被降低,而不是被消除,导致测定中的萤光素酶信号的下降。carrière等人突出显示了导致报告信号下降的可能原因,例如完整噬菌体-表达的基因和测定的温度限制,都起源于报告噬菌体的裂解周期没被消除这一事实。依赖于噬菌体的天然溶原化周期的报告测定可预期偶发地表现出裂解活性。另外,依赖于噬菌体溶原化周期的测定可倾向于来自已经被类似噬菌体溶原化的靶细胞的超感染免疫力,以及天然存在的宿主限制系统,其靶向进来的病毒核酸,因此限制了这些报告噬菌体的宿主范围。在其它实例中,设计了转导颗粒产生系统以包装外源核酸分子,但是转导颗粒通常含有外源核酸分子和天然后代病毒核酸分子的组合。天然病毒可以表现出阻碍测定表现的裂解活性,并且病毒的裂解活性必须被消除,以便纯化转导颗粒。然而,该纯化通常是不可能的。在名称为reporterplasmidpackagingsystemfordetectionofbacteria(用于检测细菌的报告质粒包装系统)的u.s.2009/0155768a中,scholl等人描述了这类转导颗粒系统的开发。该系统的产物是报告转导颗粒和天然噬菌体的组合(参考图8)。尽管作者指出转导颗粒和天然噬菌体可以通过超速离心来分离,该分离仅在以下系统中才是可能的:在所述系统中,转导颗粒和天然病毒表现出允许通过超速离心来分离的不同密度。尽管参考文献中所述的基于噬菌体t7的包装系统表现出该特征,但是这并非是在其它病毒系统中通常可用的特征。对于病毒包装机器表现出头容纳量包装是常见的,这将导致天然病毒和转导颗粒表现出无法通过超速离心来分离的不能区分的密度。病毒包装系统也依赖于合适病毒结构装配所需的最小量的包装,这导致天然病毒和转导颗粒具有不能区分的密度。因此,需要这样的非复制型转导颗粒:其没有来自病毒裂解功能的有害效应和被超感染免疫力和宿主限制机制(其靶向病毒核酸分子和病毒功能)限制的可能性,所有这些都可通过增加检测限和导致假阴性结果而限制报告测定的表现。甚至当转导颗粒被工程改造时,使用转导颗粒来检测和报告细胞中靶核酸分子的存在的方法也有限制。一些方法需要破坏细胞和麻烦的技术以分离和检测裂解物中的转录本。检测方法包括使用带标记的探针,例如抗体、适体或核酸探针。靶向靶基因的带标记的探针可导致与无意的靶标的非特异性结合或产生具有高信噪比的信号。因此,需要特异性的、有效而精确的方法来检测和报告细胞中的内源核酸分子。因此,需要产生非复制型转导颗粒的方法和系统,其允许细胞中的报告分子的包装和表达,同时消除复制型后代病毒。也需要使用表达的报告分子来检测细胞中的分子的有效而准确的方法。发明概述本文公开的是用于将报告核酸分子包装入非复制型转导颗粒的细菌细胞包装系统,所述细菌细胞包含:溶原化的噬菌体基因组,其缺乏编码包装起始位点序列的噬菌体基因,其中所述噬菌体基因的缺失阻止将噬菌体核酸分子包装入所述非复制型转导颗粒;和报告核酸分子,其包含第二噬菌体基因,其中所述第二噬菌体基因编码包装起始位点序列并促使将所述报告核酸分子的复制物包装入所述非复制型转导颗粒,其中所述第二噬菌体基因能够表达所述基因编码的蛋白,其中所述报告核酸分子的所述复制物形成复制子,其易于被包装入所述非复制型转导颗粒。在某些实施方案中,所述报告核酸分子与启动子操作性连接。在另一个实施方案中,选择所述启动子,以有助于所述报告核酸分子在所述细菌细胞中表达的报告分子的反应性。在一个实施方案中,所述报告核酸分子包含复制起点。在再一个实施方案中,所述复制子包括多联体(concatamer),其易于被包装入所述非复制型转导颗粒。在一个实施方案中,所述第一和所述第二噬菌体基因各自包括肠杆菌科噬菌体p1的paca基因并包含所述包装起始位点序列。在一个实施方案中,所述第二噬菌体基因包含seqidno:9序列。在另一个实施方案中,所述复制子是肠杆菌科噬菌体p1裂解复制子。在某些实施方案中,所述复制子包括c1阻遏物-控制的p53启动子、启动子p53反义、repl基因和kila基因的框内缺失。在一个实施方案中,所述复制子包含seqidno:3序列。在再一个实施方案中,所述第一和所述第二噬菌体基因各自包括小末端酶(ters)基因,其包含所述包装起始位点序列。在一个实施方案中,所述ters基因是金黄色葡萄球菌噬菌体φ11或φ80αters基因。在另一个实施方案中,所述复制子源自金黄色葡萄球菌pt181质粒复制起点。在再一个实施方案中,所述复制子包含seqidno:5序列。在某些实施方案中,所述第二噬菌体基因的包装起始位点序列包含pac-位点。在其它实施方案中,所述第二噬菌体基因的pac-位点包含seqidno:7序列。一方面,所述第二噬菌体基因的包装起始位点序列包含cos-位点。另一方面,所述第二噬菌体基因的包装起始位点序列包含多联体连接。另一方面,质粒包含所述报告核酸分子。一方面,第二噬菌体基因与启动子操作性连接。在另一个实施方案中,所述启动子是诱导型启动子或组成型启动子。在一个实施方案中,所述噬菌体包含肠杆菌科噬菌体p1。在再一个实施方案中,所述噬菌体包括金黄色葡萄球菌噬菌体φ80α或噬菌体φ11。一方面,所述细菌细胞包含大肠杆菌细胞。另一方面,所述细菌细胞包含金黄色葡萄球菌细胞。在再一个实施方案中,所述细菌细胞包括革兰氏阴性细胞。在其它实施方案中,所述细菌细胞包括革兰氏阳性细胞。另一方面,所述报告核酸分子包括报告基因。一方面,所述报告基因编码可检测的和/或可选择的标记。在某些方面,所述报告基因选自介导发光反应的酶(luxa、luxb、luxab、luc、ruc、nluc)、介导比色反应的酶(lacz、hrp)、荧光蛋白(gfp、egfp、yfp、rfp、cfp、bfp、mcherry、近红外荧光蛋白)、亲和肽(his-标签、3x-flag)和可选择的标记(ampc、tet(m)、cat、erm)。另一方面,所述报告核酸分子包含适体。再一方面,所述报告核酸分子包括核酸转录本序列,其与所述报告核酸分子中的第二序列互补。在一个实施方案中,所述核酸转录本序列与细胞转录本互补。在另一个实施方案中,所述核酸转录本序列包含顺式阻遏序列。在再一个实施方案中,所述报告核酸分子的复制物包括核酸转录本序列,其与所述报告核酸分子的所述复制物中的第二序列互补,其中所述核酸转录本序列与细胞转录本互补并且其中所述核酸转录本序列包含顺式阻遏序列。在某些实施方案中,将报告核酸分子包装入非复制型转导颗粒的方法,包括提供条件给本文所述的细菌细胞,所述条件诱导所述噬菌体的裂解期以产生用所述报告核酸分子包装的非复制型转导颗粒;和分离所述非复制型转导颗粒,其包含所述报告核酸分子。在一个实施方案中,所述非复制型转导颗粒不含复制的噬菌体基因组。在另一个实施方案中,对所述裂解期的诱导触发所述基因组岛核酸分子从所述细菌细胞的所述基因组中切除。在另一个实施方案中,包含所述非复制型转导颗粒的组合物包含根据本文所述的方法产生的所述报告核酸分子的复制物。本发明包括用于将报告核酸分子包装入非复制型转导颗粒的细菌细胞包装系统,所述细菌细胞包含:溶原化的噬菌体基因组,其包含第一噬菌体包装起始位点序列,其中所述第一噬菌体包装起始位点序列包含阻止将噬菌体核酸分子包装入所述非复制型转导颗粒的突变;和报告核酸分子,其包含第二噬菌体包装起始位点序列,其中所述第二噬菌体包装起始位点序列缺乏所述突变并促使将所述报告核酸分子的复制物包装入所述非复制型转导颗粒,其中所述报告核酸分子的所述复制物形成复制子,用于包装入所述非复制型转导颗粒。在一个实施方案中,所述报告核酸分子与启动子操作性连接。在另一个实施方案中,选择所述启动子,以有助于所述报告核酸分子在所述细菌细胞中表达的报告分子的反应性。在再一个实施方案中,所述报告核酸分子包含复制起点。在一个实施方案中,所述复制子包括多联体,其易于被包装入所述非复制型转导颗粒。另一方面,所述第一和所述第二噬菌体包装起始位点序列各自包含来自小末端酶基因的包装起始位点序列。一方面,所述第一和所述第二噬菌体包装起始位点序列各自包含来自肠杆菌科噬菌体p1的paca基因的pac-位点序列。另一方面,所述第一噬菌体包装起始位点序列包含seqidno:2。再一方面,所述第二噬菌体包装起始位点序列包含seqidno:1。在一个实施方案中,所述复制子包含肠杆菌科噬菌体p1裂解复制子。在另一个实施方案中,所述复制子包括c1阻遏物-控制的p53启动子、启动子p53反义、repl基因和kila基因的框内缺失。另一方面,所述复制子包含seqidno:3序列。在某些方面,所述第一和所述第二噬菌体包装起始位点序列各自包含来自金黄色葡萄球菌噬菌体φ11或φ80α的小末端酶(ters)基因的pac-位点序列。另一方面,所述复制子源自金黄色葡萄球菌pt181质粒复制起点。再一方面,所述复制子包含seqidno:5序列。一方面,所述第一噬菌体包装起始位点序列包含seqidno:2序列。在某些实施方案中,所述第二噬菌体包装起始位点序列包含seqidno:1序列。在其它实施方案中,所述包装起始位点序列包含pac-位点。在另一个实施方案中,所述包装起始位点序列包含cos-位点。在再一个实施方案中,所述包装起始位点序列包含多联体连接。在某些实施方案中,所述第一噬菌体包装起始位点序列中的所述突变包含沉默突变。在另一个实施方案中,所述第一噬菌体包装起始位点序列中的所述突变阻止所述包装起始序列的切割。在另一个实施方案中,质粒包含所述报告核酸分子。在一个实施方案中,所述噬菌体包含肠杆菌科噬菌体p1。在另一个实施方案中,所述噬菌体包含金黄色葡萄球菌噬菌体φ11或φ80α。在一个实施方案中,所述细菌细胞包含大肠杆菌细胞。在另一个实施方案中,所述细菌细胞包含金黄色葡萄球菌细胞。在某些实施方案中,所述细菌细胞包括革兰氏阴性细菌细胞。一方面,所述细菌细胞包括革兰氏阳性细菌细胞。另一方面,所述报告核酸分子包括报告基因。再一方面,所述报告基因编码可检测的标记和/或可选择的标记。在其它方面,所述报告基因选自:编码介导发光反应的酶的基因(luxa、luxb、luxab、luc、ruc、nluc),编码介导比色反应的酶的基因(lacz、hrp),编码荧光蛋白(gfp、egfp、yfp、rfp、cfp、bfp、mcherry、近红外荧光蛋白)的基因,编码亲和肽(his-标签、3x-flag)的核酸分子,和编码可选择的标记(ampc、tet(m)、cat、erm)的基因。另一方面,所述报告核酸分子包含适体。在其它方面,所述复制子被噬菌体包装机器包装入所述非复制型转导颗粒。在某些实施方案中,所述报告核酸分子包括核酸转录本序列,其与所述报告核酸分子中的第二序列互补。在另一个实施方案中,所述核酸转录本序列与细胞转录本互补。一方面,所述核酸转录本序列包含顺式阻遏序列。另一方面,所述报告核酸分子的复制物包括核酸转录本序列,其与所述报告核酸分子的所述复制物中的第二序列互补,其中所述核酸转录本序列与细胞转录本互补,和其中所述核酸转录本序列包含顺式阻遏序列。在某些方面,将报告核酸分子包装入非复制型转导颗粒的方法包括:提供条件给本文所述的细菌细胞,所述条件诱导所述噬菌体的裂解期以产生用所述报告核酸分子包装的非复制型转导颗粒;和分离所述非复制型转导颗粒,其包含所述报告核酸分子。在其它方面,所述非复制型转导颗粒不含复制的噬菌体基因组。一方面,对所述裂解期的诱导触发所述基因组岛核酸分子从所述细菌细胞的所述基因组中切除。另一方面,本发明包括包含所述非复制型转导颗粒的组合物,所述非复制型转导颗粒包含根据本文所述的方法产生的所述报告核酸分子的复制物。一方面,本发明包括用于将报告核酸分子包装入非复制型转导颗粒的细菌细胞包装系统,所述细菌细胞包含:溶原化的噬菌体基因组,其包含含有所述第一噬菌体基因的包装起始位点序列的缺失的第一噬菌体基因,所述缺失阻止将噬菌体核酸分子包装入所述非复制型转导颗粒;和报告核酸分子,其包含第二噬菌体基因,所述第二噬菌体基因包含促使将所述报告核酸分子的复制物包装入所述非复制型转导颗粒的第二包装起始位点序列,其中所述第二噬菌体基因编码蛋白,其中所述报告核酸分子的所述复制物形成复制子,用于包装入所述非复制型转导颗粒。另一方面,所述报告核酸分子与启动子操作性连接。一方面,选择所述启动子,以有助于所述报告核酸分子在所述细菌细胞中表达的报告分子的反应性。在某些方面,所述报告核酸包含复制起点。另一方面,所述复制子包括多联体,其易于被包装入所述非复制型转导颗粒。一方面,所述第一和所述第二噬菌体基因各自包括肠杆菌科噬菌体p1的paca基因并包含所述包装起始位点序列。另一方面,第一噬菌体基因包含seqidno:6序列。在某些方面,第二噬菌体基因包含seqidno:7序列。一方面,所述复制子包含肠杆菌科噬菌体p1裂解复制子。再一方面,所述复制子包含c1阻遏物-控制的p53启动子、启动子p53反义、repl基因和kila基因的框内缺失。另一方面,所述复制子包含seqidno:3序列。在其它方面,所述第一和所述第二噬菌体基因各自包括小末端酶(ters)基因,其包含所述包装起始位点序列。一方面,所述ters基因是金黄色葡萄球菌噬菌体φ11或φ80αters基因。另一方面,所述第一噬菌体基因包含seqidno:8序列。再一方面,所述第二噬菌体基因包含seqidno:9序列。一方面,所述复制子源自金黄色葡萄球菌pt181质粒复制起点。在一个实施方案中,所述复制子包含seqidno:5序列,在另一个实施方案中,所述第二噬菌体基因的包装起始位点序列包含pac-位点。在再一个实施方案中,所述第二噬菌体基因的包装起始位点序列包括cos-位点。在某些实施方案中,所述第二噬菌体基因的包装起始位点序列包含多联体连接。在一个实施方案中,质粒包含所述报告核酸分子。在另一个实施方案中,第二噬菌体基因与启动子操作性连接。在再一个实施方案中,所述启动子是诱导型启动子或组成型启动子。在某些实施方案中,所述噬菌体包含肠杆菌科噬菌体p1。在一个实施方案中,所述噬菌体包含金黄色葡萄球菌噬菌体φ80α或噬菌体φ11。在其它实施方案中,所述细菌细胞包含大肠杆菌细胞。在另一个实施方案中,所述细菌细胞包含金黄色葡萄球菌细胞。在一个实施方案中,所述细菌细胞包括革兰氏阴性细胞。在另一个实施方案中,所述细菌细胞包括革兰氏阳性细胞。另一方面,所述报告核酸分子包括报告基因。一方面,所述报告基因编码可检测的和/或可选择的标记。另一方面,所述报告基因选自:编码介导发光反应的酶(luxa、luxb、luxab、luc、ruc、nluc)的基因,编码介导比色反应的酶(lacz、hrp)的基因,编码荧光蛋白(gfp、egfp、yfp、rfp、cfp、bfp、mcherry、近红外荧光蛋白)的基因,编码亲和肽(his-标签、3x-flag)的核酸分子,和编码可选择的标记(ampc、tet(m)、cat、erm)的基因。在一个实施方案中,所述报告核酸分子包含适体。在另一个实施方案中,所述复制子被噬菌体包装机器包装入所述非复制型转导颗粒。在再一个实施方案中,所述报告核酸分子包括核酸转录本序列,其与所述报告核酸分子中的第二序列互补。在一个实施方案中,所述核酸转录本序列与细胞转录本互补。在另一个实施方案中,所述核酸转录本序列包含顺式阻遏序列。在某些实施方案中,所述报告核酸分子的复制物包括核酸转录本序列,其与所述报告核酸分子中的所述复制物的第二序列互补,其中所述核酸转录本序列与细胞转录本互补和其中所述核酸转录本序列包含顺式阻遏序列。本发明包括用于将报告核酸分子包装入非复制型转导颗粒的方法,包括:提供条件给权利要求86-125中任一项的所述细菌细胞,所述条件诱导所述噬菌体的裂解期以产生用所述报告核酸分子包装的非复制型转导颗粒;和分离所述非复制型转导颗粒,其包含所述报告核酸分子。在一个实施方案中,所述非复制型转导颗粒不含复制的噬菌体基因组。在另一个实施方案中,对所述裂解期的诱导触发所述基因组岛核酸分子从所述细菌细胞的所述基因组中切除。在一些方面,本发明包括包含所述非复制型转导颗粒的组合物,所述非复制型转导颗粒包含根据本文所述的方法产生的所述报告核酸分子的复制物。另一方面,本发明包括用于将报告核酸分子包装入非复制型转导颗粒的细菌细胞包装系统,所述细菌细胞包含:溶原化的噬菌体基因组,其缺乏包装基因并包含编码形成所述非复制型转导颗粒的蛋白的基因;和基因组岛核酸分子,其包含报告核酸分子和包装基因。一方面,所述包装基因包括小末端酶(ters)基因。ters基因包括金黄色葡萄球菌噬菌体φ80αters基因或噬菌体φ11ters基因。一方面,所述ters基因包含seqidno:9序列。另一方面,所述基因组岛核酸分子包括sapibov2基因组岛核酸分子。再一方面,所述基因组岛核酸分子选自sapi、sapi1、sapi2、sapibov1和sapibov2基因组岛核酸分子。在另一个实施方案中,所述报告核酸分子与启动子操作性连接。在再一个实施方案中,所述报告核酸分子包含复制起点。在某些实施方案中,所述噬菌体包括金黄色葡萄球菌噬菌体φ80α或噬菌体φ11。在其它实施方案中,所述细菌细胞包含金黄色葡萄球菌细胞。在一个实施方案中,所述基因组岛核酸分子包含整合酶基因和其中所述整合酶基因编码整合酶蛋白,用于将所述基因组岛核酸分子从所述细菌细胞的细菌基因组上切除掉或用于将所述基因组岛核酸分子整合入所述细菌细胞的细菌基因组。在另一个实施方案中,所述整合酶基因包含seqidno:10序列。在再一个实施方案中,所述基因组岛核酸分子被整合入所述细菌细胞的细菌基因组。在某些方面,所述基因组岛核酸分子可以被复制并形成分子复制子,其易于被噬菌体包装机器包装入所述细菌细胞。另一方面,所述核酸分子形成多联体。再一方面,所述复制的基因组岛核酸分子能够被包装入所述非复制型转导颗粒。在某些方面,所述包装基因包括pac位点序列。另一方面,所述包装基因包括cos-位点序列。在再一个实施方案中,所述包装基因包括多联体连接。在其它实施方案中,所述报告核酸分子包括报告基因。在某些实施方案中,所述报告基因编码可选择的标记和/或可选择的标记。在另一个实施方案中,所述报告基因选自介导发光反应的酶(luxa、luxb、luxab、luc、ruc、nluc)、介导比色反应的酶(lacz、hrp)、荧光蛋白(gfp、egfp、yfp、rfp、cfp、bfp、mcherry、近红外荧光蛋白)、亲和肽(his-标签、3x-flag)和可选择的标记(ampc、tet(m)、cat、erm)。在某些实施方案中,所述报告核酸分子包含适体。在其它实施方案中,所述基因组岛核酸分子缺乏整合酶基因。在另一个实施方案中,本发明包括细菌基因,其包含操作性连接到启动子的整合酶基因和其中所述整合酶基因编码整合酶蛋白,用于将所述基因组岛核酸分子从所述细菌细胞的细菌基因组上切除掉或将所述基因组岛核酸分子整合入所述细菌细胞的细菌基因组。在一个实施方案中,所述报告核酸分子包括核酸转录本序列,其与所述报告核酸分子中的第二序列互补。在其它实施方案中,所述核酸转录本序列与细胞转录本互补。在再一些实施方案中,所述核酸转录本序列包含顺式阻遏序列。在另一个实施方案中,所述报告核酸分子的复制物包括核酸转录本序列,其与所述报告核酸分子的所述复制物中的第二序列互补。在其它实施方案中,所述核酸转录本序列与细胞转录本互补。在其它实施方案中,所述核酸转录本序列包含顺式阻遏序列。本发明包括用于将报告核酸分子包装入非复制型转导颗粒的方法,包括:提供条件给权利要求130-160中任一项的所述细菌细胞,所述条件诱导所述噬菌体的裂解期以产生用所述报告核酸分子包装的非复制型转导颗粒;和分离所述非复制型转导颗粒,其包含所述报告核酸分子。在某些实施方案中,所述非复制型转导颗粒不含复制的噬菌体基因组。在一个实施方案中,对所述裂解期的诱导触发所述基因组岛核酸分子从所述细菌细胞的所述基因组中切除。在另一个实施方案中,本发明包括包含所述非复制型转导颗粒的组合物,所述非复制型转导颗粒包含根据本文所述的方法产生的所述报告核酸分子的复制物。本发明也包括用于检测样品中细菌细胞的存在或不存在的方法,包括:在使得所述非复制型转导颗粒可以转导所述细菌细胞和其中所述报告基因可以在所述细菌细胞中表达的条件下,将非复制型转导颗粒引入样品,所述非复制型转导颗粒包含编码报告分子的报告基因和缺乏噬菌体基因组;提供所述报告分子活化的条件;和检测从所述表达的报告分子传送的报告信号的存在或不存在,其中所述报告信号的存在准确地指示所述细菌细胞的所述存在。在一个实施方案中,所述方法达到相对于标准的至少80%检测特异性、相对于标准的至少90%检测特异性、或相对于标准的至少95%检测特异性。在另一个实施方案中,所述方法达到相对于标准的至少80%检测灵敏性、相对于标准的至少85%检测灵敏性、或相对于标准的至少90%检测灵敏性、或相对于标准的至少95%检测灵敏性。在再一个实施方案中,所述方法达到相对于标准的至少95%检测特异性和至少90%检测灵敏性。在另一个实施方案中,所述标准是金标准。在再一个实施方案中,所述细菌细胞包括甲氧西林耐药性金黄色葡萄球菌(mrsa)细胞。在其它实施方案中,所述细菌细胞包括甲氧西林敏感性金黄色葡萄球菌(mssa)细胞。在另一个实施方案中,所述报告基因编码可检测的或可选择的标记。在一个实施方案中,所述报告基因选自:编码介导发光反应的酶(luxa、luxb、luxab、luc、ruc、nluc)的基因,编码介导比色反应的酶(lacz、hrp)的基因,编码荧光蛋白(gfp、egfp、yfp、rfp、cfp、bfp、mcherry、近红外荧光蛋白)的基因,编码亲和肽(his-标签、3x-flag)的核酸分子,和编码可选择的标记(ampc、tet(m)、cat、erm)的基因。在一个实施方案中,所述报告基因与组成型启动子操作性连接。另一方面,从样品中可检测所述报告信号的检测限(lod)为小于1,000菌落形成单位(cfu)。在其它方面,从样品中可检测所述报告信号的检测限(lod)为小于100菌落形成单位(cfu)。一方面,从样品中可检测所述报告信号的检测限(lod)为小于10菌落形成单位(cfu)。在其它方面,从样品中可检测所述报告信号的lod为小于5cfu。另一方面,从样品中可检测所述报告信号的lod为3或更小cfu。在一个实施方案中,所述方法包括向所述样品提供预定浓度的抗生素和检测所述报告信号的存在或不存在以测定所述细菌细胞是否对所述抗生素耐药或敏感。在另一个实施方案中,所述方法包括向所述样品提供不同预定浓度的抗生素和检测所述报告信号的量以确定所述细菌细胞对所述抗生素的最小抑制浓度。一方面,本发明包括组合物,其包含编码核酸报告转录本的核酸构建体,所述核酸报告转录本能够形成包括以下的至少两种构象:阻止报告基因表达的第一构象,所述第一构象包含含有第一亚序列和第二亚序列的分子内双链区;和缺乏所述分子内双链区和允许报告基因表达的第二构象,其中所述第一和第二构象之间的转化是由细胞转录本与所述第一和/或所述第二亚序列的竞争性结合而介导。另一方面,本发明包括包含所述核酸构建体的非复制型转导颗粒。再一方面,所述细胞转录本与所述第一和/或所述第二亚序列的所述竞争性结合导致所述核酸报告构建体的所述第二构象。一方面,所述第一亚序列或所述第二亚序列包含顺式阻遏序列。另一方面,所述顺式阻遏序列包含与所述细胞转录本的一部分互补或基本互补的序列。在其它方面,所述第一亚序列或所述第二亚序列包含报告基因序列。再一方面,所述报告基因序列包含核糖体结合位点。在其它方面,所述报告基因序列编码可检测的分子。另一方面,所述可检测的标记包括荧光分子或能够介导发光或比色反应的酶。在一个实施方案中,所述报告基因序列编码可选择的标记。在另一个实施方案中,所述可选择的标记包含抗生素耐药性基因。在其它实施方案中,所述第一亚序列和所述第二亚序列彼此顺式地位于所述核酸构建体上,形成所述分子内双链区。在某些实施方案中,所述第一亚序列和所述第二亚序列彼此互补或基本互补,形成所述分子内双链区。在一个实施方案中,所述第一构象的第一亚序列或所述第二亚序列包括转录增强子序列,和其中所述转录增强子序列在所述报告基因序列的编码区的上游。在另一个实施方案中,所述核酸报告转录本的第一构象能够与切割酶结合。在其它实施方案中,所述核酸报告转录本的第一构象是细胞酶所降解的靶标。在其它方面,所述第一构象包括非结合分子内区。另一方面,所述非结合分子内区位于所述第一亚序列的3’和所述第二亚序列的5’。在其它方面,所述非结合分子内区包含序列yunr,其中y是嘧啶,u是尿嘧啶,n是任何核苷酸,r是嘌呤。在一个实施方案中,所述第一亚序列或所述第二亚序列包含所述细胞转录本的修饰的序列。在另一个实施方案中,所述修饰的序列包含核苷酸取代。在再一个实施方案中,所述修饰的序列包含序列插入、所述细胞转录本的缺失或倒转。所述方法包括组合物,其包含:编码核酸报告转录本的核酸构建体,所述核酸报告转录本包含基因报告序列并且能够形成以下的至少两种构象的所述核酸报告转录本:阻止所述核酸报告转录本中的所述报告基因序列的翻译的第一不稳定的构象,和所述第一不稳定的构象与细胞转录本结合所致的第二稳定的构象,所述第二稳定的二级构象允许所述核酸报告转录本的所述报告基因序列的翻译。在一个实施方案中,所述组合物包括包含所述核酸构建体的非复制型转导颗粒。在另一个实施方案中,所述细胞转录本结合在所述核酸报告转录本的3’utr序列。在一个实施方案中,所述第二稳定的二级构象通过所述第一不稳定的二级构象的一部分序列的切割而形成。在另一个实施方案中,所述报告基因序列编码可检测的分子。在某些实施方案中,所述可检测的标记包括荧光分子或能够介导发光或比色反应的酶。在其它实施方案中,所述报告基因序列编码可选择的标记。在另一个实施方案中,所述可选择的标记包含抗生素耐药性基因。本发明也包括组合物,其包含:编码核酸报告转录本的核酸构建体,所述核酸报告转录本包含报告基因序列并且能够形成包括以下的至少两种构象的所述核酸报告转录本:阻止所述核酸构建体进一步转录的第一构象,和在所述第一构象与细胞转录本结合后形成的第二构象,其中所述第二构象允许所述核酸构建体转录。在某些实施方案中,所述组合物包括包含所述核酸构建体的非复制型转导颗粒。在另一个实施方案中,所述核酸报告转录本包含顺式阻遏序列。在一个实施方案中,所述核酸报告转录本包含报告基因序列。在另一个实施方案中,所述第一构象由所述顺式阻遏序列与所述报告基因序列的结合而形成。在某些实施方案中,所述第一构象是切割酶的底物。在一个实施方案中,所述核酸报告转录本的第一构象包含形成转录终止结构的序列。在其它实施方案中,所述细胞转录本与所述形成转录终止结构的序列的结合导致所述核酸报告转录本的一部分的切割和所述第二构象的形成。本发明包括载体,其包含与编码本文所述的核酸报告转录本的核酸序列操作性连接的调节序列。本发明包括用于在细胞中检测靶转录本的方法,包括:将本文所述的核酸报告构建体引入所述细胞;和检测来自所述细胞的输出信号的存在或不存在,其中所述输出信号的所述存在指示所述细胞中的靶转录本的存在。所述方法包括根据检测所述靶转录本的所述存在来检测细菌细胞的存在。在一个实施方案中,检测样品中细菌细胞的存在的方法包括将本文所述的核酸报告构建体引入所述样品;和检测来自所述样品的输出信号的存在或不存在,其中所述输出信号的所述存在指示所述样品中的细菌细胞的存在。本发明包括试剂盒,其包含:容纳包含细胞的样品和本文所述的核酸报告构建体的隔间;和用于检测来自所述样品的输出信号的存在或不存在的说明书,其中输出信号的存在指示所述细胞中的靶转录本的存在。本发明包括组合物,其包含:含有核酸报告构建体的非复制型转导颗粒,所述核酸报告构建体包含与报告基因操作性连接的第一启动子,其中所述第一启动子能够被细菌细胞中的内源性诱导蛋白所诱导。本发明包括用于检测样品中细菌细胞的存在的方法,包括:使所述样品与含有核酸报告构建体的非复制型转导颗粒接触,所述核酸报告构建体包含与报告基因操作性连接的第一启动子,其中所述第一启动子能够被所述细菌细胞中的内源性诱导蛋白所诱导;和检测来自所述报告基因的输出信号的存在或不存在,其中所述输出信号的所述存在指示所述样品中的所述细菌细胞的存在。在一个实施方案中,第一启动子与操作性连接至所述细菌细胞中的靶核酸分子的诱导型启动子相同。本发明包括组合物,其包含:含有核酸报告构建体的非复制型转导颗粒,所述核酸报告构建体包含编码报告分子的报告基因,所述非复制型转导颗粒能够进入细菌细胞;和对于所述细菌细胞而言是外源性的笼蔽的底物(cagedsubstrate),一旦被解笼蔽(un-caged),能够与所述细胞中的所述报告分子反应。本发明包括用于检测样品中细菌细胞的存在的方法,包括:使所述样品与笼蔽的底物和包含核酸报告构建体的非复制型转导颗粒接触,所述核酸报告构建体包含编码报告分子的报告基因,对于所述细胞而言是外源性的笼蔽的底物,一旦被解笼蔽,能够与所述细菌细胞中的所述报告分子结合;和检测来自所述报告分子的输出信号的存在或不存在,其中所述输出信号的所述存在指示所述样品中的所述细菌细胞的存在。在一个实施方案中,所述细胞中的靶酶与所述笼蔽的底物结合,产生解笼蔽的底物。在某些实施方案中,所述解笼蔽的底物与所述报告分子反应,产生所述输出信号。本发明也包括组合物,其包含:含有核酸报告构建体的非复制型转导颗粒,所述核酸报告构建体编码可切换的分子,所述分子能够与细菌细胞中的靶分子结合而形成复合物;和底物,所述底物能够穿透所述细胞并与所述复合物结合而产生来自所述细胞的可检测信号。本发明包括用于检测样品中细菌细胞的存在的方法,包括:使所述样品与底物和包含核酸报告构建体的非复制型转导颗粒接触,所述核酸报告构建体编码可切换的分子,所述可切换的分子能够与所述细胞中的靶分子结合而形成复合物;所述底物能够与所述复合物结合而形成底物-结合的复合物;和检测来自所述底物-结合的复合物的输出信号的存在或不存在,其中所述输出信号的所述存在指示所述样品中的所述细菌细胞的存在。在一个实施方案中,所述可切换的分子与所述靶分子的结合导致所述可切换的分子的构象变化。在另一个实施方案中,所述可切换的分子的所述构象变化允许所述底物与所述复合物结合。附图简述根据以下描述和附图,将会更好地理解本发明的这些和其它特征、方面和优势,其中:图1说明了根据本发明的实施方案,基于沉默突变/互补的p1质粒包装系统的设计和功能的实例。图2说明了根据本发明的实施方案,pgwp10001载体的示意图。图3说明了根据本发明的实施方案,pac-位点缺失/互补质粒包装系统的设计和功能的实例。图4说明了根据本发明的实施方案,pgw80a0001载体的示意图。图5描绘了根据本发明的实施方案,噬菌体的基因组岛(gi)包装的过程。图6描绘了根据本发明的实施方案,基于gi的包装系统的设计和功能的实例。图7描绘了根据本发明的实施方案,缺乏整合酶基因的基于gi的包装系统的设计和功能。图8描绘了根据本发明的实施方案,缺乏整合酶基因的基于sapibov2的包装系统的设计和功能。图9描绘了根据本发明的实施方案,使用nrtps的系统,用于检测活细胞内的靶基因启动子的诱导物。图10描绘了根据本发明的实施方案,包括经构建用于检测vanr的报告核酸分子(例如,质粒)的报告系统,vanr是屎肠球菌(enterococcusfaecium)或粪肠球菌(e.faecalis)中的万古霉素耐药性(vana)基因的启动子的诱导物。报告质粒携带报告基因,其与vana基因启动子操作性连接。图11描绘了根据本发明的实施方案,包括经构建用于检测tcdd的报告核酸分子的报告系统,tcdd是艰难梭菌(c.difficile)的毒素a和b基因(分别为tcda和tcdb)的启动子的诱导物。所述报告核酸分子包括报告基因,其与tcda基因启动子操作性连接。图12描绘了根据本发明的实施方案,包括经构建用于检测sars的报告核酸分子的报告系统,sars是金黄色葡萄球菌(s.aureus)的蛋白a基因(spa)的启动子的诱导物。所述报告核酸分子包括细菌萤光素酶基因luxa和luxb,其与spa基因启动子(pspa)操作性连接。图13显示了根据本发明的实施方案,包括用于检测活细胞内的胞内酶的系统的报告系统,其使用笼蔽的底物分子,所述底物分子可以被靶胞内酶解笼蔽。图14描绘了根据本发明的实施方案,β-内酰胺酶检测系统的设计和功能。图15显示了根据本发明的实施方案,用于检测活细胞内的胞内分子的报告系统,其使用可切换的分子,所述分子在其与靶分子结合后能够产生可检测信号。图16描绘了根据本发明的实施方案,基于噬菌体/可切换的-适体(sa)的胞内分子报告系统的设计和功能。图17描绘了根据本发明的实施方案,使用顺式阻遏机制的系统实例,其可靶向报告转录本上的报告序列的5’utr(非翻译区)。图18显示了根据本发明的实施方案,用于检测细胞中的靶转录本的存在的系统实例,其基于靶向报告转录本中的报告序列的核糖体结合位点(rbs)的顺式阻遏机制。图19说明了根据本发明的实施方案,用于检测细胞中的靶转录本的存在的示例性的系统,其基于靶向报告转录本中的报告序列的编码区(“aug”)的顺式阻遏机制。图20说明了根据本发明的实施方案,用于检测细胞中的靶转录本的存在的示例性的系统,其基于阻遏机制,使用不稳定的报告转录本。图21显示了根据本发明的实施方案,转导测定结果,其中将36种四环素-敏感性mrsa暴露给携带pgw80a0001的转导颗粒,然后点种到含有5ug/ml四环素的培养基板上。图22说明了根据本发明的实施方案,从80种mrsa的临床分离物和28种用转导颗粒转导的甲氧西林敏感性金黄色葡萄球菌(mssa)临床分离物所测定的发光。图23显示了在4、8、16、32、64和128ug/ml头孢西丁中的金黄色葡萄球菌生长结果。图24显示了在4、8、16、32、64和128ug/ml头孢西丁存在时,通过nrtp测定得到的rlu值。图24中的x轴设定在mssarlu截止值。图25显示了基于最低能量构象而产生的meca转录本的二级结构,所述最低能量构象是通过mfold而计算并用varna可视化。图26显示了meca转录本的末端环23(t23),其含有yunr共有序列。图27描绘了添加到luxab基因的5’端并经设计而形成茎-环结构的顺式阻遏序列,所述茎-环结构阻断luxa基因的rbs序列(“aaggaa”)。图28显示了靶转录本和报告转录本的顺式阻遏序列之间的碱基对的图解。图29显示了根据本发明的实施方案,靶meca基因序列的实例。图30显示了根据本发明的实施方案,示例性的meca转录本序列,其可用于设计报告转录本(seqidno:16)。图31是根据本发明的实施方案,luxab基因座dna序列的实例,其可用于设计报告转录本。图32是根据本发明的实施方案,luxab转录本序列的实例,其可用于设计报告转录本(seqidno:18)。图33是根据本发明的实施方案,luxab顺式阻遏的转录本序列的实例,其可用于报告转录本(seqidno:19)。图34显示了根据本发明的实施方案,包含载体的细胞的实例,所述载体编码报告转录本,其中在细胞中没有内源meca转录本。图35显示了引入细胞的载体,其中所述载体编码报告转录本,其包括顺式阻遏序列和报告序列(luxa基因和luxb基因)。当存在于细胞中的meca转录本与顺式阻遏序列结合时,抑制性发夹环打开并且luxa基因的rbs被暴露出来。报告序列(luxa和luxb)的翻译可以发生,导致形成luxab酶。luxab酶产生可检测的发光信号。以此方式,转录本报告载体报告了在细胞内的内源meca转录本的存在。发明详述i.定义权利要求和说明书中使用的术语定义如下,除非另有说明。本文使用的,“报告核酸分子”是指包含dna或rna分子的核苷酸序列。报告核酸分子可以是天然存在的或人工或合成的分子。在某些实施方案中,所述报告核酸分子对于宿主细胞而言是外源的并且可以作为外源核酸分子例如质粒或载体而引入宿主细胞。在某些实施方案中,所述报告核酸分子可以与细胞中的靶基因互补。在其它实施方案中,所述报告核酸分子包括编码报告分子(例如,报告酶、蛋白)的报告基因。在某些实施方案中,所述报告核酸分子被称为“报告构建体”或“核酸报告构建体”。“报告分子”或“报告物(reporter)”是指赋予生物体可检测的或可选择的表型的分子(例如,核酸或蛋白)。可检测的表型可以是例如比色的、荧光或发光的。报告分子可以是自以下的报告基因表达:编码介导发光反应的酶(luxa、luxb、luxab、luc、ruc、nluc)的基因,编码介导比色反应的酶(lacz、hrp)的基因,编码荧光蛋白(gfp、egfp、yfp、rfp、cfp、bfp、mcherry、近红外荧光蛋白)的基因,编码亲和肽(his-标签、3x-flag)的核酸分子,和编码可选择的标记(ampc、tet(m)、cat、erm)的基因。报告分子可用作核酸分子或外源序列(质粒)成功摄取入细胞的标记。报告分子也可用于指示靶基因、靶核酸分子、靶胞内分子、或细胞的存在,如本文所述。或者,所述报告分子可以是核酸,例如适体或核酶。在本发明的一些方面,所述报告核酸分子与启动子操作性连接。在本发明的其它方面,根据启动子在特定细胞(例如,特定物种)中、而非其它物种中的活性,选择或设计所述启动子,以有助于报告系统的反应性和交叉反应性。在某些方面,所述报告核酸分子包含复制起点。在其它方面,根据复制起点在特定细胞(例如,特定物种)中、而非其它物种中的活性,复制起点的选择可以类似地有助于报告系统的反应性和交叉反应性,当报告核酸分子在靶细胞内的复制有助于报告信号产生或是报告信号产生所必需时。在某些实施方案中,所述报告核酸分子形成复制子,所述复制子在病毒复制期间能够作为多联体dna被包装入后代病毒。本文使用的,“靶转录本”是指dna序列或mrna分子的核苷酸序列的一部分,其是由靶细胞天然形成的,其包括在靶基因的转录期间形成的和作为初级转录产物的rna加工产物的mrna。靶转录本也可称为细胞转录本或天然存在的转录本。本文使用的,术语“转录本”是指自dna或rna模板序列或基因转录而来的一段核苷酸序列(dna或rna)。转录本可以是自rna模板转录而来的cdna序列或自dna模板转录而来的mrna序列。转录本可以是编码蛋白的或非编码蛋白的。转录本也可自工程化核酸构建体转录而来。源自报告核酸分子的转录本可被称为“报告转录本”。报告转录本可包括报告序列和顺式阻遏序列。报告转录本可以具有形成互补区的序列,使得转录本包含形成双链体的两个区(例如,分子间双链体区)。一个区可以称为“顺式阻遏序列”并与靶转录本和/或报告序列的一部分或全部具有互补性。转录本的第二区称为“报告序列”并可与顺式阻遏序列具有互补性。互补性可以是完全互补性或基本互补性。顺式阻遏序列的存在和/或与报告序列的结合可在报告转录本中形成构象,其可以阻断报告分子的进一步表达。报告转录本可以形成二级结构,例如发夹结构,使得报告转录本内的彼此互补的区域可以彼此杂交。“引入细胞中,”当涉及核酸分子或外源序列(例如,质粒、载体、构建体)时,是指促进摄取或吸收入细胞,正如本领域技术人员所理解的。核酸构建体或转录本的吸收或摄取可通过未受协助的扩散或主动细胞过程而发生,或通过辅助剂或装置(包括通过使用噬菌体、病毒和转导颗粒)而发生。该术语的含义不限于在体外的细胞;核酸分子也可被“引入细胞”,其中所述细胞是活生物体的一部分。在这种情况下,引入细胞将包括递送到生物体。例如,对于在体内递送,可将本发明的核酸分子、构建体或载体注射入组织部位或系统性给予。体外引入细胞包括本领域已知的方法,例如电穿孔和脂质转染。进一步的方法描述于本文或是本领域已知的。“转导颗粒”是指能够将非病毒核酸分子递送入细胞的病毒。所述病毒可以是噬菌体、腺病毒等。“非复制型转导颗粒”是指能够将非病毒核酸分子递送入细胞、但是不能将其自身的复制的病毒基因组包装入转导颗粒的病毒。所述病毒可以是噬菌体、腺病毒等。“质粒”是小dna分子,其在物理上独立于细胞内染色体dna,并可独立于细胞内染色体dna而复制。最常见的是作为细菌中的小的环状、双链dna分子,质粒有时也存在于古细菌和真核生物中。质粒被认为是复制子,能够在合适宿主中自主复制。“载体”是用作运载工具而人工携带外源遗传物质进入另一细胞的核酸分子,在细胞中它可复制和/或表达。“病毒”是小的感染因子,其仅在其它生物体的活细胞内复制。病毒颗粒(产物病毒粒子)包括2或3部分:i)dna或rna分子构成的遗传物质,其携带遗传信息;ii)保护这些基因的蛋白外壳;和在某些情况下,iii)环绕蛋白外壳的脂质包膜。“mrsa”是指甲氧西林-耐药性金黄色葡萄球菌。“mssa”是指甲氧西林-敏感性金黄色葡萄球菌。术语“改善”是指导致疾病状态的治疗的任何治疗益处,包括对疾病状态的预防、减少其严重程度或进程、缓解或对其治愈。术语“原位”是指发生在活细胞中的过程,所述活细胞独立于活生物体而生长,例如生长在组织培养中。术语“在体内”是指发生在活生物体内的过程。本文使用的术语“哺乳动物”包括人和非人,并且包括但不限于人、非人类灵长类、犬类、猫类、鼠类、牛类、马类和猪类。“g,”“c,”“a”和“u”各自通常代表分别含有鸟嘌呤、胞嘧啶、腺嘌呤和尿嘧啶作为碱基的核苷酸。“t”和“dt”在本文中可互换使用,是指脱氧核糖核苷酸,其中核碱基是胸腺嘧啶,例如脱氧核糖胸腺嘧啶(deoxyribothymine)。然而,可以理解,术语“核糖核苷酸”或“核苷酸”或“脱氧核糖核苷酸”也可指修饰的核苷酸,正如以下进一步详述的那样,或替代的置换部分。技术人员完全理解鸟嘌呤、胞嘧啶、腺嘌呤和尿嘧啶可以是被其它部分置换而基本不改变寡核苷酸的碱基对性质,所述寡核苷酸包括携带这类置换部分的核苷酸。例如但不限于,包含肌苷作为其碱基的核苷酸可以与含有腺嘌呤、胞嘧啶或尿嘧啶的核苷酸碱基配对。因此,含有尿嘧啶、鸟嘌呤、或腺嘌呤的核苷酸可以在本发明的核苷酸序列中被含有例如肌苷的核苷酸置换。包含这类置换部分的序列是本发明的实施方案。本文使用的,术语“互补性”,当用于描述第一核苷酸序列与第二核苷酸序列的关系时,是指包含第一核苷酸序列的寡核苷酸或多核苷酸与包含第二核苷酸序列的寡核苷酸或多核苷酸在某些条件下杂交并形成双链体结构的能力,正如技术人员将会理解的那样。互补序列也被描述为彼此结合并通过结合亲和性来表征。例如,第一核苷酸序列可以被描述为与第二核苷酸序列互补,当这两个序列在严格性杂交条件下杂交(例如,退火)时。杂交条件包括用于退火和/或洗涤步骤的温度、离子强度、ph和有机溶剂浓度。术语严格性杂交条件是指这样的条件:在该条件下,第一核苷酸序列将优先与其靶序列例如第二核苷酸序列杂交,并且在较少程度上与其它序列杂交、或完全不与其它序列杂交的条件。严格性杂交条件是序列依赖性的,并且在不同环境参数下是不同的。通常,选择严格性杂交条件为在限定的离子强度和ph时比核苷酸序列的热融解点(tm)低大约℃。tm是50%的第一核苷酸序列与完美匹配的靶序列杂交的温度(在限定的离子强度和ph时)。核酸杂交的广泛指南参见例如,tijssen(1993)laboratorytechniquesinbiochemistryandmolecularbiology--hybridizationwithnucleicacidprobes第i部分,第2章,“overviewofprinciplesofhybridizationandthestrategyofnucleicacidprobeassays,”elsevier,n.y.(“tijssen”)。可以使用其它条件,例如生物体内可遇到的生理相关条件。根据杂交的核苷酸的最终应用,技术人员将能确定最适合两个序列互补性试验的条件组。这包括包含第一核苷酸序列的寡核苷酸或多核苷酸与包含第二核苷酸序列的寡核苷酸或多核苷酸在第一和第二核苷酸序列的全长上的碱基配对。这类序列在本文可称为彼此“完全互补性”。然而,当第一序列被认为与本文的第二序列“基本互补”时,这两个序列可以是完全互补,或它们在杂交时可形成一个或多个、但通常不超过4、3或2个错配的碱基对,同时保留在与它们的最终应用最相关的条件下杂交的能力。然而,当两个寡核苷酸经设计而在杂交时形成一个或多个单链突出端时,这样的突出端将不被认为是对于互补性测定而言的错配。例如,dsrna包含长度为21个核苷酸的一个寡核苷酸和包含长度为23个核苷酸的另一个寡核苷酸,其中较长的寡核苷酸包含与较短的寡核苷酸完全互补的21个核苷酸的序列,仍然可被认为是对于本文所述的目的的“完全互补性”。本文使用的“互补性”序列,也可包括非-watson-crick碱基对和/或自非天然的和修饰的核苷酸形成的碱基对,或完全地自所述碱基对而形成,只要其杂交能力满足上述需求。这样的非-watson-crick碱基对包括但不限于,g:uwobble或hoogstein碱基配对。本文的术语“互补性”、“完全互补性”和“基本互补性”可用于dsrna的两条链之间、或dsrna的反义链和靶序列之间、单链rna序列或单链dna序列的互补链之间的碱基匹配,正如从其使用的上下文中将会理解的那样。本文使用的,“双链体结构”包含两条反向平行的和基本互补的核酸序列。核酸构建体中的互补序列在两条转录本之间,转录本内的两个区域之间,或转录本和靶序列之间可形成“双链体结构”。一般而言,每条链的大部分核苷酸是核糖核苷酸,但是如本文详述的,每条链或两条链也可包括至少一个非核糖核苷酸,例如,脱氧核糖核苷酸和/或修饰的核苷酸。形成双链体结构的两条链可以是一个较大rna分子的不同部分,或它们可以是单独的rna分子。当两条链是一个较大分子的部分,并因此通过在一条链的3’端和形成双链体结构的相应的另一条链的5’端之间的不间断的核苷酸链而连接时,连接的rna链被称为“发夹环”。当两条链通过并非在一条链的3’端和形成双链体结构的相应的另一条链的5’端之间的不间断的核苷酸链的方式而共价连接时,连接结构被称为“接头”。rna链可以具有相同或不同数量的核苷酸。碱基对的最大数量是双链体的最短链的核苷酸数量减去双链体中存在的任何突出端。通常,双链体结构的长度介于15和30之间,或介于25和30之间,或介于18和25之间,或介于19和24之间,或介于19和21之间,或19、20或21个碱基对之间。在一个实施方案中双链体的长度为19个碱基对。在另一个实施方案中双链体的长度为21个碱基对。当两个不同的sirna组合使用时,双链体的长度可以相同或可以不同。本文使用的,术语“互补区”是指与序列例如本文定义的靶序列基本互补的反义链上的区。当互补区并非与靶序列完全互补时,在末端区的错配最能容忍,并且,如果存在的话,通常在末端区或例如在5’和/或3’端的6、5、4、3或2个核苷酸内的区域。在两个或更多个核酸或多肽序列的情况下,术语%“同一性”是指两个或更多个序列或亚序列,当对于最大一致性来比较和比对时,通过使用下述序列比较算法之一而测定(例如,blastp和blastn或技术人员可用的其它算法)或通过目测,其具有指定%的相同核苷酸或氨基酸残基。根据应用,%“同一性”可以存在于所比较的序列的区,例如功能域,或者存在于所比较的两个序列的全长。对于序列比较,通常一个序列充当参考序列,将试验序列与之比较。当使用序列比较算法时,将试验序列和参考序列输入计算机,指定亚序列坐标,必要时,并且指定序列算法程序参数。再用序列比较算法来计算试验序列相对于参考序列的%序列同一性,根据所指定的程序参数。对于比较,可进行序列的最佳比对,例如,通过smith&waterman,adv.appl.math.2:482(1981)的局部同源性算法,通过needleman&wunsch,j.mol.biol.48:443(1970)的同源性比对算法,通过pearson&lipman,proc.nat'l.acad.sci.usa85:2444(1988)的搜索相似性方法,通过这些算法的计算机执行(gap,bestfit,fasta和tfasta,wisconsingeneticssoftwarepackage,geneticscomputergroup,575sciencedr.,madison,wis.),或通过目测(一般性参见ausubel等人,下文)。适于确定%序列同一性和序列相似性的算法的一个实例是blast算法,其描述于altschul等人,j.mol.biol.215:403-410(1990)。进行blast分析的软件通过国立生物技术信息中心(nationalcenterforbiotechnologyinformation(www.ncbi.nlm.nih.gov/))是公众可得的。术语“足够量”是指足以产生所需效果的量,例如足以从细胞中产生可检测信号的量。术语“治疗有效量”是有效改善疾病症状的量。治疗有效量可以是“预防有效量”,因为预防可以被认为是治疗。必须注意,如本说明书和所附权利要求书中使用的,单数形式“一个”、“一种”和“所述”包括复数对象,除非另有明确说明。ii.病毒的溶原化和裂解周期病毒在宿主细胞中经历溶原化和裂解周期。如果采用溶原化周期,可将噬菌体染色体整合入细菌染色体,或它可以使自己成为在宿主中稳定的质粒,在宿主中,它可长时间保持静息。如果溶原性细菌被诱导,从细菌染色体上切除噬菌体基因组并启动裂解周期,这以细菌裂解和噬菌体颗粒释放而告终。裂解周期导致新噬菌体颗粒的产生,其通过宿主裂解而释放。一些温和噬菌体可以表现出裂解活性,并且其倾向性在不同宿主细菌中可以不同。为了说明该现象,经由噬斑测定检查了10个mrsa临床分离物上的两种温和金黄色葡萄球菌噬菌体的裂解活性(表1)。噬菌体φ11在10个临床mrsa分离物上有10个都表现出裂解活性并且φ80α在10个临床mrsa分离物上有6个表现出裂解活性。因此,依赖于噬菌体的天然溶原化周期的报告测定可以有望偶发地表现出裂解活性。表1:金黄色葡萄球菌温和噬菌体φ11和φ80α在10个临床mrsa分离物上的裂解活性(用字母“x”表示)mrsa分离物φ11φ80α1x2x3xx4xx5xx6x7xx8x9xx10xx另外,基于病毒的报告测定,例如基于噬菌体的报告物,可以具有有限的反应性(即分析包容性),因为由基于宿主的和前噬菌体-衍生的噬菌体抗性机制所致的在噬菌体宿主范围中的限制。这些抗性机制靶向天然噬菌体核酸,其可导致噬菌体dna和功能的降解或另外的抑制。这类抗性机制包括限制系统,其切割抑制噬菌体-衍生的转录本的噬菌体dna和crispr系统。裂解活性和噬菌体抗性对于基于报告噬菌体的测定而言都可以是抑制性的。裂解活性可以通过破坏或另外抑制细胞产生可检测信号的能力而抑制信号并因此通过降低可检测信号的量或阻止可检测信号的产生而影响检测限。噬菌体抗性机制可以限制噬菌体的宿主范围并限制基于噬菌体的报告物的包容性,通过降低可检测信号的量或阻止可检测信号的产生而类似地影响检测限。噬菌体dna掺入报告噬菌体所致的裂解活性和噬菌体抗性,可以导致在掺入这些噬菌体报告物的测定中的假阴性结果。iii.产生非复制型转导颗粒(nrtp)的方法a.用于产生非复制型转导颗粒的基于破坏/互补的方法1)沉默突变/互补包装系统本发明包括产生nrtps的方法,使用基于沉默突变/互补的方法。该非复制型转导颗粒包装系统基于将沉默突变引入病毒基因组的组件,所述组件可被病毒包装机器识别为在病毒产生期间起始基因组包装的元件。这类元件的实例包括pac-型噬菌体的pac-位点序列和cos-型噬菌体的cos-位点序列。因为这些包装起始位点通常存在于病毒产生所必需的基因编码区内,所以将沉默突变引入,使得pac-位点不再被病毒包装机器识别为包装起始位点。同时,所述突变不破坏编码该位点的基因。通过破坏包装位点序列,突变的病毒能够经历裂解周期,但不能将其基因组dna包装入其包装单元。可将外源报告核酸分子(例如质粒dna)引入宿主细胞,所述宿主细胞已经被具有突变的包装起始位点序列的病毒基因组溶原化。外源报告核酸分子可包括天然包装起始位点序列。可将外源报告核酸分子引入细胞并在细胞中复制。当突变的病毒经历裂解周期时,表达的病毒包装机器将具有天然包装起始位点序列的外源报告核酸分子包装入病毒包装单元。病毒基因组未被包装入包装单元,因为其包装起始位点序列已经突变。在某些实施方案中,包装起始位点序列中的突变包含这样的沉默突变:其阻止包装起始序列切割,但是不破坏包含包装起始位点序列的基因产物的表达。这产生非复制型转导颗粒,例如,携带复制的外源核酸分子的病毒结构组件。这类系统的一个实例基于噬菌体p1,一种pac-型噬菌体。在一个实施方案中,将包括天然p1pac位点的质粒转化入细胞。细胞被p1前噬菌体基因组溶原化。p1前噬菌体基因组包括在p1的paca基因内编码的pac-位点序列中的沉默突变。当前噬菌体的裂解周期被诱导时,系统导致携带质粒dna的基于p1的转导颗粒的产生。适于该系统的一个沉默突变的实例描述于美国公布号2005/0118719,2002年11月7日提交,其通过引用全部结合到本文中。在seqidno:2中也发现一个实例,列举如下(p1pac-位点具有沉默突变,小写字母表示突变的碱基)。图1说明了根据本发明的实施方案,基于沉默突变/互补的p1质粒包装系统100的设计和功能的实例。在该系统中,大肠杆菌细胞101被p1前噬菌体102溶原化,所述前噬菌体102在包装起始位点序列(例如,pac-位点)包括沉默突变。细胞被含有天然pac-位点103的质粒转化,并且所述质粒在细胞中复制以形成质粒多联体104。质粒也可包括编码报告分子的报告基因。当p1前噬菌体的裂解周期被诱导时,从细菌基因组上切除p1前噬菌体并表达p1结构组件,例如衣壳蛋白105。p1结构组件仅包装含有天然pac-位点的dna(例如,质粒dna),因此产生携带质粒dna106(例如,报告基因)的非复制型转导颗粒。用于基于沉默突变/互补的p1质粒包装系统的示例性载体示于图2。有关如何构建基于沉默突变/互补的p1质粒包装系统的株系和载体的详情详述于以下实施例1。2)基于缺失/互补的包装系统本发明包括产生nrtps的方法,使用基于缺失/互补的方法。该非复制型转导颗粒包装系统基于病毒基因组的组件的缺失,所述组件可被病毒包装机器识别为在病毒产生期间起始基因组包装的元件。这类元件的实例包括pac-型噬菌体的pac-位点序列和cos-型噬菌体的cos-位点序列。这些包装起始位点通常存在于病毒产生所必需的基因编码区内。在某些实施方案中,仅缺失包装起始位点,其允许突变的病毒经历裂解周期,但不允许病毒包装其基因组dna。例如,seqidno:6是具有缺失的pac-位点序列的p1paca基因的实例(小写字母表示缺失的pac-位点序列)。在其它实施方案中,缺失包含包装起始位点的完整基因。例如,seqidno:8显示ters基因的缺失(小写字母表示缺失的序列)。在一个实例中,细胞的基因组被病毒基因组溶原化,其中包装起始位点已经缺失。将互补的质粒引入细胞,并且质粒dna包括具有包装起始位点序列的基因,其补充病毒基因组中的缺失的包装起始位点序列。当突变的病毒经历裂解周期时,病毒包装蛋白质将质粒dna的复制子包装入包装单元,因为其包装起始位点,并产生携带复制的质粒dna的非复制型转导颗粒。在某些实施方案中,优选设计缺失/互补,使得突变的病毒dna和互补的外源dna之间没有同源性。这是因为突变的病毒dna和互补的外源dna之间的同源性缺乏,避免了这两种dna分子之间同源重组的可能性,同源重组可导致将包装序列再次引入病毒基因组。为了达到同源性的缺乏,一个策略是从病毒基因组中删除含有包装起始位点序列的完整基因并随后用外源dna分子补充该基因,所述外源dna分子恰好仅含有从病毒中删除掉的dna序列。在该策略中,设计互补的dna分子以表达从病毒中删除掉的基因。使用噬菌体φ80α,一种pac-型噬菌体,提供了这类系统的另一实例。噬菌体基因组在宿主细菌细胞中溶原化,并且噬菌体基因组包括小末端酶基因,其中pac-型前噬菌体φ80α的pac-位点已经缺失。将包括互补小末端酶基因并具有天然pac-位点的质粒转化入细胞。当溶原化的前噬菌体的裂解周期被诱导时,噬菌体包装系统将质粒dna包装入后代噬菌体结构组件,而不是包装天然噬菌体dna。因此包装系统产生携带质粒dna的非复制型转导颗粒。图3说明了根据本发明的实施方案,pac-位点缺失/互补质粒包装系统300的设计和功能的实例。细菌细胞301被pac-型噬菌体302溶原化,所述噬菌体的小末端酶(ters)基因缺失。细胞被滚环复制质粒303转化,所述质粒包括补充噬菌体中的ters基因缺失的小末端酶基因。小末端酶基因含有包装起始位点序列,例如,pac-位点。质粒303也可包括编码报告分子的报告基因。包含小末端酶和大末端酶蛋白的蛋白复合物能够在pac-位点上或附近识别和切割双链dna分子,并且这允许将质粒dna分子包装入噬菌体衣壳。当细胞中的前噬菌体被诱导时,噬菌体的裂解周期产生噬菌体的结构蛋白304和噬菌体的大末端酶蛋白305。复制补充的质粒,并表达小末端酶蛋白306。将含有ters基因(和报告基因)的复制的质粒dna307包装入噬菌体衣壳,导致非复制型转导颗粒仅携带质粒dna308。图4显示了用于pac-位点缺失/互补质粒包装系统的所得载体的实例。有关pac-位点缺失/互补质粒包装系统的组件和构建的进一步的详情见以下实施例2。b.基于毒力岛的包装系统毒力岛(pathogenicityisland,ptis)是水平转移的遗传元件(称为基因组岛)的亚类。在金黄色葡萄球菌中存在着高度移动的ptis的特定家族,其被某些定居的前噬菌体诱导而切割和复制。将这些ptis包装入小头部噬菌体-样颗粒并以相当于噬菌体的噬斑形成滴度的频率转移。该过程称为sapi切除复制-包装(erp)周期,并且高频率sapi转移称为sapi-特异性转移(spst),以将其与传统的普遍性转导(cgt)区分开。sapis具有高度保守的遗传组成,其类似于噬菌体的遗传组成并且将其与所有其它水平获得的基因组岛明确区分开。sapi1-编码的和sapibov2-编码的整合酶对于相应元件的切除和整合而言是必需的,并且假定对于其它sapis同样如此。噬菌体80α可以诱导若干不同的sapis,包括sapi1、sapi2和sapibov1,而φ11可以诱导sapibov1,但不能诱导其它两种sapis。图5描绘了噬菌体对基因组岛(gi)包装500的天然过程。事实上,被合适的前噬菌体503溶原化并携带gi504的细菌细胞501可产生携带gi多联体512的噬菌体颗粒。在该过程中,当噬菌体被诱导进入其裂解周期时,从细菌基因组502上切除(未显示)噬菌体基因组,该噬菌体基因组随后表达噬菌体蛋白,包括衣壳成分505和大末端酶蛋白(terl)506。前噬菌体诱导也经由gi整合酶蛋白(int)507的表达而触发gi切除。以切除的噬菌体基因组的类似方式(未显示),gi环化(circularizes)508,表达其自身的小末端酶蛋白(ters)509并开始复制形成gi多联体510。然后噬菌体terl基因和giters基因可经由gi基因组中的pac-位点序列而组合、结合并切割gi多联体,和随后可将gi多联体包装入噬菌体衣壳511,产生携带gi多联体512的噬菌体颗粒。在天然系统中,如图5所示,自噬菌体产生的所得裂解物同时包括天然噬菌体颗粒、以及含有gi的噬菌体颗粒。天然噬菌体颗粒是天然噬菌体基因组包装的结果,因为噬菌体基因组多联体内的pac-位点的识别。1)基因组岛(gi)包装系统设计和功能本发明的产生nrtps的方法包括基于gi的包装系统。相比质粒包装系统,天然gi-包装系统得益于以下事实:被包装的dna源自细菌基因组内的基因组区并因此无需细菌宿主维持质粒。在某些实施方案中,本发明包括细菌细胞包装系统,用于将报告核酸分子包装入非复制型转导颗粒,其中所述细菌细胞包括缺乏包装基因的溶原化的噬菌体基因组,和基因组岛、隐蔽性噬菌体、或其它核酸分子(需要噬菌体例如heper噬菌体来动员该核酸分子),并包括报告核酸分子和包装基因。基于基因组岛的系统可以基于例如金黄色葡萄球菌毒力岛(sapis)、大肠杆菌criptic噬菌体p4和辅助噬菌体p2、和肠球菌criptic噬菌体p7和辅助噬菌体p1。可以利用gi-包装系统,使得噬菌体包装外源核酸序列。这可以通过将这类外源核酸序列掺入gi而完成。为了从该过程中消除天然噬菌体,前噬菌体的小末端酶基因可以缺失。小末端酶基因序列含有天然噬菌体的pac-位点序列,该缺失具有阻止天然噬菌体dna包装的作用。在其它实施方案中,仅小末端酶基因的pac位点可以缺失。将被包装的gi包括其自身的pac-位点和表达合适的小末端酶蛋白的小末端酶基因,并且仅gidna将会被包装入该系统。图6描绘了根据本发明的实施方案,基于gi的包装系统600的设计和功能的实例。在该系统中,细菌细胞601的基因组被合适的前噬菌体603溶原化,前噬菌体的小末端酶基因缺失,和细胞的基因组602携带gi604。当噬菌体被诱导进入其裂解周期时,从细菌基因组602上切除噬菌体基因组(未显示)。噬菌体基因组表达噬菌体蛋白,包括衣壳成分605和大末端酶蛋白(terl)606。前噬菌体诱导也经由gi整合酶蛋白(int)607的表达而触发gi切除。以切除的噬菌体基因组的类似方式(未显示),gi环化608和表达其自身的小末端酶蛋白(ters)609并且经复制形成gi多联体610。然后噬菌体terl基因和giters基因可经由gidna中的pac-位点序列而组合、结合并切割gi多联体。随后可将gi多联体包装入噬菌体衣壳611,产生携带gi多联体612的噬菌体颗粒。在该系统中,噬菌体dna将不会被包装入噬菌体颗粒,因为它缺乏含有噬菌体的pac-位点序列的ters基因,并因此而不能被表达的giters和噬菌体terl蛋白识别。当将含有包装的gidna的噬菌体颗粒给予受体细胞时,噬菌体将结合到受体细胞表面,然后将包装的gidna多联体引入细胞。一旦在细胞内,gi可以再次表达其整合酶蛋白,然后gi可整合入受体细胞基因组的其特定位点。在包装前,如果外源dna序列包括在gi中时,包装系统因此允许将外源dna序列递送到受体细胞并将这些外源dna序列整合入受体细胞基因组。2)缺乏整合酶的基于gi的包装系统在另一个实施方案中,设计上述包装系统,使得包装的gidna不能整合入受体细胞基因组。这可以通过缺失gi中的整合酶基因并通过导致与gi呈反式的整合酶基因的表达补充该缺失而实现。以此方式,对于包装宿主细胞中的gi的切除而言,整合酶蛋白是可用的,并且已被包装入噬菌体的gidna不含整合酶基因且不能表达整合酶蛋白,因此阻止了所递送的gi的整合。图7描绘了根据本发明的实施方案,缺乏int基因700的基于gi的包装系统的设计和功能。在该系统中,细菌细胞701被合适的前噬菌体溶原化,所述前噬菌体的小末端酶基因缺失703。细胞的基因组702携带gi,其具有整合酶(int)基因缺失704并且也携带与合适的启动子705操作性连接的缺失的int基因。因此int基因可表达与gi706呈反式的整合酶蛋白(int)。当噬菌体被诱导进入裂解周期,从细菌基因组702上切除噬菌体基因组(未显示),该噬菌体基因组随后表达噬菌体蛋白,包括衣壳成分707和大末端酶蛋白(terl)708。前噬菌体诱导也经由整合酶蛋白707的表达而触发gi切除。以切除的噬菌体基因组的类似方式(未显示),切除的gi环化709,表达其自身的小末端酶蛋白(ters)710,并开始复制形成gi多联体711。然后噬菌体terl基因和giters基因可经由gidna中的pac-位点序列而组合、结合并切割gi多联体,随后可将gi多联体包装入噬菌体衣壳712,产生携带gi多联体713的噬菌体颗粒。在该系统中,噬菌体dna将不会被包装,因为它缺乏含有噬菌体的pac-位点序列的ters基因,并因此而不能被表达的giters和噬菌体terl蛋白识别。当将含有包装的gidna(缺乏int基因)的噬菌体颗粒给予受体细胞时,噬菌体将结合到受体细胞表面,然后将包装的gidna多联体引入细胞。一旦在细胞内,gi因为缺乏整合酶基因而不能表达其整合酶蛋白,然后gi不能整合入受体细胞基因组的其特定位点。在包装前,如果外源dna序列包括在gi中时,因此包装系统允许将外源dna序列递送到受体细胞,并且所递送的dna序列不在用于gi整合的特定位点整合入受体细胞基因组。3)缺乏整合酶的基于sapibov2的包装的设计和功能在某些实施方案中,产生nrtps的方法采用在基于gi的包装系统中的gisapibov2和噬菌体φ11。替代实施方案可以采用其它sapigi’s和其它合适的噬菌体,包括sapi’ssapi1、sapi2、sapibov1和sapibov2连同噬菌体80α,和sapi’ssapibov1和sapibov2连同噬菌体φ11。根据以下描述,本领域技术人员将会知道如何开发不缺乏int基因的基于gi的包装系统,正如部分iia中所述。图8描绘了根据本发明的实施方案,缺乏int基因的基于sapibov2的包装系统800的设计和功能。在该系统中,金黄色葡萄球菌细胞801被φ11溶原化,其具有小末端酶基因缺失803。细胞的基因组802携带sapibov2,其具有其整合酶(int)基因缺失804并且也携带缺失的int基因,该基因与组成型表达的pclpb基因启动子805操作性连接。int基因可表达与sapibov2806呈反式的整合酶蛋白(int)。当噬菌体被诱导进入其裂解周期时,从细菌基因组802上切除噬菌体基因组(未显示),该噬菌体基因组随后表达噬菌体蛋白,包括衣壳成分807和大末端酶蛋白(terl)808。前噬菌体诱导经由整合酶蛋白806的表达也触发了sapibov2切除。以切除的噬菌体基因组的类似方式(未显示),切除的sapibov2环化809,表达其自身的小末端酶蛋白(ters)810并开始复制形成sapibov2多联体811。然后噬菌体terl基因和sapibov2ters基因可经由sapibov2dna中的pac-位点序列而组合、结合并切割sapibov2多联体,随后可将sapibov2多联体包装入噬菌体衣壳812,产生携带812多联体813的噬菌体颗粒。在该系统中,噬菌体dna将不会被包装,因为它缺乏含有噬菌体的pac-位点序列的ters基因,并因此而不能被表达的sapibov2ters和噬菌体terl蛋白识别。iv.报告物在某些实施方案中,本发明的nrtps和构建体包含报告核酸分子,包括报告基因。报告基因可编码报告分子,和报告分子可以是可检测的或可选择的标记。在某些实施方案中,报告基因编码报告分子,当报告分子在细胞中表达时产生可检测信号。在某些实施方案中,所述报告分子可以是荧光报告分子,例如但不限于,绿色荧光蛋白(gfp)、增强的gfp、黄色荧光蛋白(yfp)、青色荧光蛋白(cfp)、蓝色荧光蛋白(bfp)、红色荧光蛋白(rfp)或mcherry、以及近红外荧光蛋白。在其它实施方案中,所述报告分子可以是介导发光反应的酶(luxa、luxb、luxab、luc、ruc、nluc等)。报告分子可包括细菌萤光素酶、真核萤光素酶、适于比色检测的酶(lacz、hrp)、适于免疫检测的蛋白例如亲和肽(his-标签、3x-flag)、充当适体或表现出酶活性的核酸(核酶),或可选择的标记、例如抗生素耐药性基因(ampc、tet(m)、cat、erm)。本领域已知的其它报告分子可用于产生信号,以检测靶核酸或细胞。在其它方面,所述报告分子包括核酸分子。在一些方面,所述报告分子是具有特异性结合活性或表现出酶活性(例如,aptazyme、dnazyme、核酶)的适体。在本文的部分v进一步描述了报告物和报告测定。v.nrtps和报告测定a.诱导物报告测定本发明包含使用nrtps作为报告分子的方法,与靶向活细胞内基因启动子的内源的或天然的诱导物一起使用。可使用部分iii和以下实施例1-6中所述的方法,将本发明的nrtps工程化。在某些实施方案中,所述方法包含使用nrtp作为报告物,其中nrtp包括与诱导型启动子操作性连接的报告基因,所述启动子控制靶细胞内的靶基因的表达。当将包含报告基因的nrtp引入靶细胞时,经由报告核酸分子中的靶基因启动子的诱导,报告基因的表达是可能的。图9描绘了具有以下两个基因的靶细胞900的基因组座:一个基因编码诱导物902和一个靶基因903。也描绘的是报告核酸分子904,其包括与靶细胞的靶基因的启动子906操作性连接的报告基因905。经由nrtp,可将报告核酸分子904引入细胞。在天然细胞中,当诱导物基因902被表达和产生诱导蛋白907时,诱导蛋白907能够诱导与靶基因操作性连接的靶基因启动子906,因此导致靶基因的表达和靶基因产物908的产生。当报告核酸分子904存在于靶生物中时,诱导物907也能诱导报告核酸分子904内存在的靶基因启动子906,因此导致报告基因905的表达,导致报告分子909的产生,其能够产生可检测信号。因此,根据靶细胞内诱导蛋白907的存在,自报告分子909产生可检测信号指示细胞的存在。1)vanr报告系统在一个实施方案中,所述报告系统包括nrtp,其包含报告核酸分子(例如,质粒)。可构建报告核酸分子,用于检测vanr,vanr是屎肠球菌或粪肠球菌中的万古霉素耐药性(vana)基因的启动子的诱导物。报告质粒携带报告基因,其与vana基因启动子操作性连接。图10概述了vanr报告系统的设计和功能。图10描绘了转座子tn15461001的区域,其可存在于屎肠球菌中。tn1546转座子可包括vanr诱导物基因1002和vana靶基因1003。图中也描绘的是报告核酸分子1004,其可以被包装入nrtp并引入细胞。报告核酸分子1004包括报告基因1005,其与启动子ph1006操作性连接,该启动子控制包括vana基因在内的vanhax操纵子的表达。在天然细胞中,当vanr基因1002被表达和产生vanr蛋白1007时,vanr能够诱导tn1546转座子中的ph1006,因此导致vana基因的表达和vana蛋白1008的产生。当报告核酸分子1003(载体)存在于靶生物中时,vanr也能诱导报告核酸分子1003内的ph1006,因此导致报告分子1009的表达。因此,报告分子的产生指示靶细胞内的vanr的存在。适于开发vre测定的启动子的实例包括:vana基因启动子和vanb基因启动子。arthur,m.,等人,thevanssensornegativelycontrolsvanr-mediatedtranscriptionalactivationofglycopeptideresistancegenesoftn1546andrelatedelementsintheabsenceofinduction(在诱导不存在时,vans传感器负面地控制vanr-介导的tn1546和相关元件的糖肽抗性基因的转录活化).j.bacteriol.,1997.179(1):第97-106页。2)tcdd报告系统在该系统的另一实施方案中,使用nrtp,将报告核酸分子引入细胞。可构建报告核酸分子,用于检测tcdd,tcdd是艰难梭菌的毒素a和b基因(分别为tcda和tcdb)的启动子的诱导物。所述报告核酸分子包括报告基因,其与tcda基因启动子操作性连接。图11概述了根据本发明的实施方案,tcdd报告系统的设计和功能。图11描绘了转座子paloc1101的区域,其可存在于艰难梭菌中。paloc转座子可含有tcdd基因1102和tcda靶基因1103。图中也描绘的是报告核酸分子1104(例如,载体),使用nrtp,将其引入细胞。报告核酸分子1104包括报告基因1105,其与tcda基因启动子(ptcda)1106操作性连接。在天然细胞中,当tcdd基因被表达和产生tcdd蛋白1107时,tcdd能够诱导paloc转座子1101中的ptcda1106,因此导致tcda基因1103的表达和毒素a蛋白1108的产生。当报告核酸分子1104存在于靶生物中时,tcdd也能诱导报告载体内的ptcda1106,因此导致报告分子1109的表达。因此,报告分子1109的产生指示靶细胞内的tcdd的存在。适于开发艰难梭菌测定的启动子的实例包括:tcda基因启动子和tcdb基因启动子。karlsson,s.,等人,expressionofclostridiumdifficiletoxinsaandbandtheirsigmafactortcddiscontrolledbytemperature(艰难梭菌毒素a和b及其sigmafactortcdd的表达受到温度的控制).infect.immun.,2003.71(4):第1784-1793页。靶细胞和诱导物:靶细胞可包括真核细胞靶和原核细胞靶和相关诱导物。载体递送系统:含有重组dna的载体的递送可以通过非生物或生物系统来进行。包括但不限于脂质体、病毒-样颗粒、衍生自噬菌体或病毒的转导颗粒、和接合。3)基于噬菌体的sars报告系统在本发明的另一实施方案中,构建报告核酸分子,用于检测sars,sars是金黄色葡萄球菌的蛋白a基因(spa)的启动子的诱导物。在nrtp中,可将报告核酸分子引入细胞并且包括细菌萤光素酶基因luxa和luxb,其与spa基因启动子(pspa)操作性连接。经由例如nrtp,将报告核酸分子递送到金黄色葡萄球菌。如果sars存在于细胞中,它将诱导luxab基因的表达,因此产生萤光素酶,其能够产生发光信号。图12概述了根据本发明的一个实施方案,sars报告系统的设计和功能。图12描绘了金黄色葡萄球菌基因组1201的区域,其含有sars基因1202和spa基因1203。图中也描绘的是通过nrtp递送给细胞的报告核酸分子(例如,载体)1204,并且其包括luxab报告基因1205,所述基因与控制spa基因1203的表达的启动子pspa1206操作性连接。在天然细胞中,当sars基因1202被表达,产生sars蛋白1207时,该蛋白能够诱导金黄色葡萄球菌基因组转座子中的pspa1206,因此导致spa基因1203的表达和蛋白a1208的产生。当报告核酸分子1204存在于靶生物中时,sars1207也能诱导报告核酸分子1204内的pspa1206,因此导致luxab的表达,导致萤光素酶1209的产生,其可以产生发光信号。因此,萤光素酶的产生指示靶细胞内的sars的存在。b.酶报告测定根据本发明的实施方案,本发明包括用于检测活细胞内的胞内酶的系统,其使用笼蔽的底物分子,所述底物分子可以被靶胞内酶解笼蔽。图13描绘了胞内酶检测系统的设计和功能。用nrtp(未显示),将报告分子-表达载体1301递送到靶细胞1302。经由nrtp,报告分子-表达载体1301能够穿透靶细胞1302并将报告分子基因1303递送入靶细胞1302,然后报告分子1304可自报告分子基因1303而表达。也将笼蔽的底物1305加入靶细胞1302中并且其能够穿透入靶细胞1302。如果靶胞内酶1307存在于靶细胞1306中,酶1307能够去除笼蔽的底物1305的笼蔽组件,因此产生解笼蔽的底物1308。解笼蔽的底物1308随后可以与细胞1302内的报告分子1304反应,该反应产物产生可检测信号1309。靶细胞和酶:靶细胞可包括真核细胞靶和原核细胞靶和相关酶,包括例如,金黄色葡萄球菌中的β-内酰胺酶。载体递送系统:含有重组dna的载体的递送可以通过非生物或生物系统来进行。包括但不限于脂质体、病毒-样颗粒、衍生自噬菌体或病毒的转导颗粒、和接合。报告分子和笼蔽的底物:可以使用多种报告分子和笼蔽的底物,正如以下文献所述的那些:danielsobek,j.r.,enzymedetectionsystemwithcagedsubstrates(用笼蔽的底物的酶检测系统),2007,zymera,inc。1)基于噬菌体的β-内酰胺酶报告物在一个实施方案中,报告分子-表达载体可以由nrtp携带,使得可将该载体递送入细菌细胞。所表达的报告分子可以是海肾萤光素酶(renillaluciferase),并且笼蔽的底物可以是被笼蔽的海肾荧光素,使得对于靶细胞而言是内源性的β-内酰胺酶能够从笼蔽的荧光素中切割笼蔽的化合物并释放解笼蔽的荧光素。图14描绘了根据本发明的实施方案,β-内酰胺酶检测系统的设计和功能。将基于噬菌体的nrtp1401所携带的海肾萤光素酶-表达载体加入靶金黄色葡萄球菌细胞1402。使用包含海肾萤光素酶-表达载体的nrtp,该载体能够穿透靶细胞1402。nrtp将海肾萤光素酶基因1403递送入靶细胞1402,随后可以自其基因表达海肾萤光素酶1404。也将笼蔽的海肾荧光素1405加入靶细胞1402并且其能够穿透入靶细胞1402。如果胞内β-内酰胺酶1407存在于靶细胞1402中,该酶能够去除笼蔽的荧光素1406的笼蔽组件,因此产生解笼蔽的荧光素1408。解笼蔽的荧光素1408随后可以与细胞1402内的海肾萤光素酶1404反应,该反应的产物导致发光1409。以此方式,当含有β-内酰胺酶的靶细胞暴露给nrtp和笼蔽的荧光素时,细胞将表现出发光信号,其指示细胞中存在的β-内酰胺酶的存在。c.胞内分子报告物本发明包括用于检测活细胞内的胞内分子的系统,其使用可切换的分子,所述可切换的分子在其与靶分子结合后能够产生可检测信号。图15描绘了基于可切换的分子(sm)的胞内分子检测系统的设计和功能。在nrtp中,将sm-表达载体1501递送到靶细胞1502。sm-表达载体1501能够穿透靶细胞1502并将sm基因1503递送入靶细胞1502。然后sm蛋白1504可自sm基因1503而表达。sm蛋白1504随后可以结合到细胞内的靶分子1505并因此形成sm-靶分子复合物1506。sm1504与靶分子1505的结合导致sm1504的构象变化,这使得结合的sm能够结合底物。将底物1508加入细胞1507并且其能够穿透入细胞1502。细胞1502内的结合的sm能够也结合底物,因此形成sm-靶分子-底物复合物1509。最终,靶分子-结合的sm与底物1508的结合具有产生可检测信号1510的作用。因此该系统所产生的可检测信号指示细胞内的靶分子的存在。靶细胞和分子:可使用多种真核和原核细胞靶并且可设计基于可切换的适体的sm,以靶向多种基于核酸和氨基酸的胞内分子靶,正如以下文献所述:samiejaffrey,j.p.,coupledrecognition/detectionsystemforinvivoandinvitrouse(用于体内和体外使用的偶联识别/检测系统),2010,cornelluniversity。载体递送系统:含有重组dna的载体的递送可以通过非生物或生物系统来进行。包括但不限于脂质体、病毒-样颗粒、衍生自噬菌体或病毒的转导颗粒、和接合。1)基于非复制型转导颗粒/可切换的适体的胞内分子报告系统在该方法的一个实例中,可切换的分子-表达载体可以被基于噬菌体的转导颗粒所携带,使得可将该载体递送入细菌细胞。所表达的可切换的分子可以是可切换的适体,其经设计而在与胞内靶分子结合后经历构象变化。构象变化允许该适体随后结合荧光团,所述荧光团当与适体结合后表现出增强的荧光。图16描绘了基于噬菌体/可切换的-适体(sa)-的胞内分子报告系统的设计和功能。将nrtp1601所携带的sa-表达载体加入靶细胞1602。nrtp1601能够将sa-表达载体和sa-表达基因1603递送入靶细胞1602。随后可自sa基因1603表达sa蛋白1604。sa蛋白1604随后可以结合到细胞内的靶分子1605并因此形成sa-靶分子复合物1606。sa1604与靶分子1605的结合导致sa的构象变化,这使得结合的sa能够结合荧光团1608。将荧光团1607加入细胞并且其能够穿透入细胞1608。细胞内的结合的sa也能够结合荧光团,因此形成sa-靶分子-荧光团复合物1609。最终,靶分子-结合的sa与荧光团的结合具有增强荧光团1610的荧光的作用。因此该系统所产生的可检测的荧光信号指示细胞内的靶分子的存在。d.转录本报告测定本发明包括包含基于反义rna的方法的报告测定,用于通过导致报告分子的表达(如果靶转录本存在于细胞内)而检测活细胞内的靶转录本。本领域的用于抑制基因表达的某些胞内方法采用小干扰rna例如双链rna(dsrna),以靶向细胞中的转录的基因。dsrna包含反义链和有义链,其被递送入细胞或在细胞中表达,并且dsrna的链经由反式作用抑制机制起作用,其中一条链(通常是反义链)与靶基因序列(rna转录本)结合并阻止靶基因序列的表达。双链rna分子已被证明以高度保守的调节机制(称为rna干扰(rnai))阻断(敲减)基因表达。wo99/32619(fire等人)公开了长度至少为25个核苷酸的dsrna在秀丽新小杆线虫(c.elegans)中抑制基因表达的用途。dsrna也被证明在其它生物体中降解靶rna,所述生物体包括植物(参见例如wo99/53050,waterhouse等人;和wo99/61631,heifetz等人)、果蝇属(drosophila)(参见例如yang,d.,等人,curr.biol.(2000)10:1191-1200)、和哺乳动物(参见wo00/44895,limmer;和de10100586.5,kreutzer等人)。然而,dsrna的一条链与靶基因的结合可以是非特异性的。如果类似机制用于检测系统的话,该非特异性结合可以导致高假阳性率,这使其不适于开发临床有用的检测系统。先前的反式作用抑制机制已被证明不适于开发临床有用的检测系统。例如,一些方法导致高水平的非特异性信号和至多90%假阳性率,当达到90%测定灵敏性时。参见美国专利号8,329,889。已经开发了用于基因表达的转录后调节的某些方法,其使用顺式阻遏的标记转录本,例如绿色荧光蛋白标记,其中该标记的核糖体结合位点被顺式阻遏序列、连同反式活化的rna转录本而阻断。当反式活化的rna转录本与顺式阻遏的标记转录本结合时,顺式阻遏的标记转录本的发夹结构被改变,标记基因的上游核糖体结合位点被暴露出来,允许标记基因的转录和表达。然而,这些方法先前未用于检测内源转录本,也没有超出控制细胞内基因表达的基本转换机制的成功。1)核酸分子相互作用和机制本发明的方法利用细胞中的转录本-水平调节机制,包括反义rna(asrna)机制,将核酸分子递送入细胞。反义机制包括所有形式的序列-特异性mrna识别,导致靶转录本表达的减少、消除、活化或其它改变。参见good,l.,translationrepressionbyantisensesequences(反义序列所致的翻译阻遏).cellularandmolecularlifesciences,2003.60(5):第854-861页,和lioliou,e.,rna-mediatedregulationinbacteria:fromnaturaltoartificialsystems(在细菌中的rna-介导的调节:从天然到人工系统),newbiotechnology.2010.27(3):第222-235页。天然存在的asrnas可见于所有3类生物界,并且它们影响信使rna(mrna)破坏、阻遏和活化,以及rna加工和转录。参见sabine,b.,antisense-rnaregulationandrnainterference(反义-rna调节和rna干扰).biochimicaetbiophysicaacta(bba)–genestructureandexpression,2001.1575(1-3):第15-25页。该机制已被用于抑制蛋白合成,用于治疗应用。反义rna是单链rna,其与细胞内转录的信使rna(mrna)链互补。可将asrna引入细胞,通过对它的碱基配对和物理上阻碍翻译机制而抑制互补性mrna的翻译。将反义rna退火到互补性mrna靶序列,并且作为或核糖体进入或核糖体通读的位阻作用的结果,中断了mrna靶序列的翻译。反义rna机制不同于rna干扰(rnai),这是一种相关过程,其中双链rna片段(dsrna,也称为小干扰rnas(sirnas))触发催化性地介导的基因沉默,最典型地通过靶向rna-诱导的沉默复合物(risc)与mrna结合并使之降解。将dsrna分子的一条链退火到mrna或dna,可通过细胞中的核糖核酸酶、或通过反义化合物本身所致的靶rna切割,而导致双链体rna、杂合rna/dna双链体或双链体rna类似前体trna的快速降解。rnai途径在许多真核生物中都发现,是由酶dicer起始,该酶将长双链rna(dsrna)分子切割为~20个核苷酸的短双链片段,其称为sirnas。每个sirna被松开成为两条单链rnas(ssrna),称为过客链(passengerstrand)和引导链(guidestrand)。过客链被降解,而引导链掺入rna-诱导的沉默复合物(risc)。在转录后基因沉默中,引导链与信使rna分子中的互补序列碱基配对,并且由称为argonaute的蛋白(risc复合物的催化组件)诱导切割。对于本发明机制的核酸相互作用,报告转录本和靶转录本之间的相互作用可以依赖于两个转录本中都存在的环之间(例如,“接吻复合物(kissingcomplex)”)、或环和单链(ss)区之间的碱基配对。在一些情况下,接吻复合物形成足以介导所需的相互作用的效应,而在另一些情况下,初步接触的蔓延将导致相互作用,产生所需效应。2)通过转录本-水平调节,报告构建体的翻译的顺式阻遏和反式活化的机制根据本发明的实施方案,基于可以使用的多种阻遏/活化机制,以下描述说明了转录本报告系统。在图17-20的各图中,载体包括包含报告序列的报告构建体,和报告构建体上的各区域示于各图,包括被顺式阻遏序列的阻遏所靶向的区域。以下描述提供了多种抑制机制(包括转录衰减、翻译衰减、和转录本的失稳定)和多种活化机制(包括构象变化和切割)的非限制性实例。图17描绘了系统1700的实例,其使用顺式阻遏机制,其可靶向报告转录本1703上的报告序列1702的5’utr(非翻译区)1701。也显示了报告序列1702(5’utr(1701)、rbs、编码区和3’utr)内的区域。顺式阻遏序列1705是报告序列和直到报告序列的5'utr1701的上游。rna聚合酶1704自载体1706转录报告构建体1703的序列。在转录期间的某一点,因为转录的顺式阻遏序列1705内的相互作用,通过在报告转录本1703中形成转录终止(tt)茎-环结构1707而终止转录过程。转录终止1707结构阻止1708rna聚合酶1704转录载体1706。在某些实施方案中,转录终止蛋白(例如,大肠杆菌中的nusa)与rna聚合酶和/或与转录终止1707结构结合,停止报告构建体的转录。当靶转录本1709存在于细胞中时,靶转录本1709与报告转录本1703结合。在某些实施方案中,靶转录本和报告转录本之间的结合是通过各自序列中的核苷酸的碱基配对。靶转录本1709和报告转录本103之间的相互作用导致转录终止(tt)茎-环结构1707被切割1710。报告转录本1703的切割可以通过细胞酶(例如rna酶iii)而发生。在这种情况下,分析靶转录本的二级结构,分析在二级结构的ssrna区中的rna酶iii共有序列的存在,例如5’-nnwawgnnnuun-3’或5’-nagnnnncwuwnn-3’,其中“n”和“n”是任何核苷酸,“w”是a或u并且“n”指示对于watson–crick碱基配对的相对严格的需求,而“n”指示碱基配对的最低需求。当这样的共有序列存在于靶转录本上时,可将转录终止结构1707的环设计为与所述rna酶iii共有序列互补,使得当在各rna分子中的ssrna杂交时,形成rna酶iii切割位点,允许切割转录终止结构1707。在meca转录本中,环t23,起始于核苷酸1,404,具有序列cagauaacauuuu,其适用于此类方法。在某些实施方案中,将切割位点工程化在报告构建体中,使得在转录后切割报告转录本。该切割,在所提供的实例中,可以发生在转录终止子结构中的紧邻环的位置。由rna聚合酶104重新启动转录1711。转录终止(tt)茎-环结构1707的切割允许报告序列1702的剩余部分的转录和随后翻译。这导致自所翻译的报告分子产生可检测的或可选择的标记。在原核生物中,转录终止结构1707包括rho-非依赖性机制,其具有长度为7-20碱基对的茎-环结构,富含胞嘧啶-鸟嘌呤碱基对并且后接尿嘧啶残基链。nusa与转录终止茎-环结构1707结合,导致在聚-尿嘧啶序列转录期间rna聚合酶停止。弱的腺嘌呤-尿嘧啶键降低了rna-dna双链体不稳定的能量,允许其从rna聚合酶上解开并脱离。在真核细胞中,转录终止结构1707被蛋白因子识别并且包括新转录本的切割,接着是聚腺苷酸化。图18显示了用于检测细胞中的靶转录本的存在的系统1800的实例,其基于靶向报告转录本1703中的报告序列1702的核糖体结合位点(rbs)1801的顺式阻遏机制。rbs1801是mrna序列,当起始蛋白翻译时,核糖体1802与该mrna序列结合。顺式阻遏序列1705被设计为与rbs1801结合(例如,顺式阻遏序列1705与rbs序列1801互补)。rbs1801与顺式阻遏序列1705结合并变为螯合的(sequestered)(核糖体1802难以接近),阻止报告转录本1703的翻译。当来自细胞的靶转录本109与报告转录本1703结合时,靶转录本1709对rbs序列1801具有较高结合亲和力,和构象变化发生在报告转录本1703中,其方式是释放顺式阻遏1705序列与rbs序列1801之间的结合。这允许核糖体1802与rbs1801结合,从而允许报告转录本1703的翻译。图19说明了用于检测细胞中的靶转录本的存在的示例性的系统1900,其基于靶向报告转录本1703中的报告序列1702的编码区(“aug”)1901的顺式阻遏机制。构建顺式阻遏序列1705,使得它与报告序列1702的编码区1901结合(例如互补结合)。“aug”起始密码子已显示是编码区1901的一部分。顺式阻遏序列1705与编码区1901的结合导致这样的构象,其导致报告构建体1703的切割1902。报告转录本1703的切割阻止了翻译。当靶转录本1709存在于细胞中时,靶转录本1709与顺式阻遏序列1705结合,其方式导致报告转录本1703中的构象变化。该构象变化阻止或去除了顺式阻遏序列1705与报告序列1702的编码区1901之间的相互作用,从而允许报告序列1702的翻译。图20说明了用于检测细胞中的靶转录本的存在的示例性的系统2000,其基于阻遏机制,使用不稳定的报告转录本2001。将报告转录本2001设计为不稳定的,使得它形成不稳定的构象,该构象阻止报告转录本2001的翻译。报告转录本2001被定义为不稳定的,如果它因多种因素(例如外来体复合物或降解体)而倾向于快速降解的话。细胞中的靶转录本1709与一部分不稳定的报告转录本2001结合。在该实例中,负责使转录本不稳定的部分位于报告序列的3’utr2005中,并且3’utr2005如同报告构建体1703的顺式阻遏序列起作用。靶转录本1709与报告序列的3’utr2005的结合导致切割事件2003,其使报告转录本2001稳定并允许报告转录本2001的翻译2004。切割发生在靶转录本1709结合后并且起到移除负责使转录本不稳定的序列部分的作用。在该实例中,靶转录本1709与报告序列的3'utr405结合,但是系统400也可被设计,使得结合和切割发生在5'utr、5'utr的上游、或3'utr的下游。结合和切割可以发生在报告序列1702的翻译所需的区域之外任何地方。在某些实施方案中,顺式阻遏序列本身包含两个序列,其可彼此结合(例如,彼此互补),并且顺式阻遏序列的这两个序列的结合所导致的报告转录本的构象阻止了报告转录本中的报告序列的翻译。3)对于阻遏/活化机制,天然存在的和合成的系统已经描述了若干天然存在的和合成产生的转录本-水平机制,证明了图17-20中说明的每个实例中使用的各个机制(即构象变化和切割)。在反义rna(asrna)-介导的转录衰减中已经观察到转录终止。在一个实例中,rnaiii/reprmrna之间的两个环–环相互作用之后形成稳定的双链体。该复合物使rho-非依赖性终止子结构稳定,以阻止由rna聚合酶(rnap)所致的延伸。经由合成的核糖开关系统的开发,已经描述了rbs螯合(sequestration)机制。在该系统中,与rbs互补的序列位于rbs上游,允许两个区之间的接头序列的存在。mrna转录后,这两个互补区杂交,产生发夹,其阻止核糖体的泊靠(docking)。为了活化翻译,携带rbs序列的合成的反式活化的rna与杂交的rna结合,允许将rbs暴露并可用于翻译。也已经在天然系统中描述了因rna切割所致的对翻译的阻止,在所述天然系统中asrnamicc靶向ompdmrna的编码区内的序列。hfq所促进的相互作用,导致rna酶e切割mrna。再一个天然机制表明切割事件活化翻译、而非抑制翻译。大肠杆菌gadyasrna靶向gadxw操纵子的两个基因之间的基因间的区域。在gady和gadx的3’utr之间形成稳定的螺旋后,rna酶切割发生在转录本中并使gadx转录本稳定,允许其翻译。4)通过报告序列的顺式阻遏和通过靶转录本的结合,构象变化的机制本发明中使用的通用机制是分子间的核酸分子相互作用,其可导致两个后续机制:(1)核酸分子的二级结构中的构象变化,和(2)切割事件。本文描述的是设计报告转录本的方法,其可经历顺式阻遏的构象和脱-阻遏的构象之间的构象变化,使得通过靶转录本与报告转录本的结合而诱导构象变化。如上所述,报告转录本可包含报告序列并且可被设计,使得报告基因序列的翻译被报告基因的核糖体结合位点(rbs)的顺式阻遏所阻断。在某些实施方案中,以下工具可以用于设计本发明的报告转录本。1)使用二级结构程序来计算rna二级结构,所述程序例如在由以下机构维持的服务器上可得的mfold:rnainstitutecollegeofartsandsciences,universityatalbany,stateuniversityofnewyork(mfold网络服务器,用于核酸折叠和杂交预测.nucleicacidsres.31(13),3406-15,(2003))(http://mfold.rna.albany.edu/q=mfold/rna-folding-form)。2)使用软件程序来计算分子间的rna相互作用,所述程序例如rna-rnainteractionprediction,使用由以下机构维持的服务器上可得的integerprogramming(ractip):graduateschoolofinformationscience,narainstituteofscienceandtechnology(naist),departmentofbiosciencesandinformatics,keiouniversityjapan(http://rna.naist.jp/ractip/)。3)使用visualizationappletforrna(varna)(http://varna.lri.fr/)使rna二级结构可视化,所述软件是用于绘制rna二级结构的javalightweightapplet。根据通过mfold而计算并用varna可视化的最低能量构象,可以产生靶转录本的二级结构。可在靶转录本内鉴定ssrna区或靶区,其对于与报告转录本的结合而言可以是理想的。在一些情况下,靶转录本的二级结构包含共有序列或环序列,其可与报告序列的一部分结合。例如,在甲氧西林-耐药性金黄色葡萄球菌的meca转录本中,具有包括共有yunr序列(“uugg”)的末端环,其可用于与报告转录本的顺式阻遏序列结合。对靶转录本的二级结构的分析可以揭示这一个或多个ssrna区,其可适于与顺式阻遏序列结合。报告转录本的顺式阻遏序列随后可以经设计而与这一个或多个ssrna区结合。在某些实施方案中,所述顺式阻遏序列可以经设计而与报告转录本中的报告序列的rbs结合并在报告转录本内形成茎-环结构,使得顺式阻遏序列阻断rna聚合酶与报告序列的rbs结合。一旦顺式阻遏序列与靶转录本的ssrna区结合,报告序列的rbs可以被暴露并且可以启动报告序列的翻译。在某些实施方案中,报告转录本的顺式阻遏序列可以经设计而位于报告序列的5’端并且经设计而在报告序列中产生茎-环结构,使得报告序列的rbs序列被阻断。根据报告转录本的最低能量构象,顺式阻遏茎-环结构可以经设计而阻断rbs序列,所述最低能量构象是通过mfold而计算并用varna可视化。靶转录本和报告转录本的顺式阻遏序列之间的预测的分子间相互作用可通过ractip而计算并通过varna可视化。可以绘制图解,使靶转录本和报告转录本的顺式阻遏序列之间的碱基配对可视化,如下图28所示。相互作用可包括靶序列和顺式阻遏序列中的5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49或50或更多个核苷酸之间的碱基配对。这两个序列之间的互补结合可以是完全互补、基本互补或部分互补。碱基配对可以是跨越靶和顺式阻遏序列内的连续的核苷酸序列或区域,例如,如图28所示。5)顺式阻遏的转录本或报告转录本的切割机制本发明中使用的通用机制是分子间的核酸分子相互作用,其可导致两个后续机制:(1)核酸分子的二级结构中的构象变化,和(2)切割事件。本文描述的是用于设计报告转录本的方法和系统,其采用切割事件。在某些实施方案中,切割机制可以用于本发明的系统和方法,用于顺式阻遏或反式活化。例如,如以上图17、19和20中所述,系统可以经设计而利用切割机制,即通过将报告转录本的核酸序列暴露给切割酶(rna酶)或螯合被序列特异性rna酶识别的单链序列。在一个实例中,在报告转录本中可设计核糖核酸酶e(rna酶e)位点(“*”指示切割位点):(g,a)n(c,a)n(g)(g,u,a)*(a,u)(c,u)n(c,a)(c,a)。参见kaberdin等人,probingthesubstratespecificityofe.colirnaseeusinganoveloligonucleotide-basedassay(使用新的基于寡核苷酸的测定,探测大肠杆菌rna酶e的底物特异性).nucleicacidsresearch,2003,第31卷,第16期(doi:10.1093/nar/gkg690)。在顺式阻遏系统中,可将顺式阻遏序列并入报告转录本的设计中,使得当转录时,报告转录本的构象暴露了在待切割的所需位点上含有序列rna酶e识别基序的单链区。在某些实施方案中,切割位点可以涉及报告转录本的转录的阻遏,例如,如果切割位点在报告基因的编码区内的话。对于反式-脱阻遏系统,顺式阻遏的转录本可以经工程化而与靶转录本结合,使得相互作用导致报告转录本的构象变化,其螯合了含有rna酶e位点的单链区。所述系统可以经设计,使得顺式阻遏机制起因于由顺式阻遏序列的构象所产生的特定二级结构,例如上述转录终止结构。在该实例中,切割事件用于使报告序列脱阻遏。这可以通过以下来达到:通过设计顺式阻遏序列,使其与天然存在的质粒或其它细胞转录本相互作用(与之结合),使得相互作用导致含有rna酶e位点的单链区的产生,该位点可被切割并因此从报告转录本上去除顺式阻遏序列。在某些实施方案中,当切割事件用于报告物的表达时,rna酶e位点被设计在报告序列的编码区之外,其在5’和3’utr中具有足够的序列长度,以便允许有活力的报告转录本。在这种情况下,rna酶e位点被设计为在原核系统中的起始密码子上游的至少0、1、2、3、4、5、10、20、30、40、50、60、70、80、90、100、200、300、400、500或更多个碱基对,和在真核系统中的起始密码子上游的至少18、20、30、40、50、60、70、80、90、100、200、300、400、500或更多个碱基对,或者在终止密码子下游的至少10、20、30、40、50、60、70、80、90、100、200、300、400、500或更多个碱基对。在其它实施方案中,当切割事件用于阻遏报告物时,rna酶e位点被设计在报告序列的编码区内或者其它位置上,以便抑制报告物的表达。6)转录本如上所述,转录本是自dna或rna模板序列或基因转录而来的一段核苷酸序列(dna或rna)。转录本可以是自rna模板转录而来的cdna序列或自dna模板转录而来的mrna序列。转录本可以是自工程化核酸构建体转录而来。转录本在其内部可以具有互补区,使得转录本包含可形成分子内双链体的两个区。一个区可以称为“顺式阻遏序列”,其与报告序列结合并阻断其翻译。转录本的第二区称为“报告序列”,其编码报告分子,例如可检测的或可选择的标记。本发明的转录本可以是长度为5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24或25个核苷酸的转录本序列。在其它实施方案中,转录本可以是长度为至少25、30、40、50、60、70、80、90、100、500、1000、1500、2000、3000、4000、5000或更多个核苷酸。顺式阻遏序列和报告序列可以是相同长度或不同长度。在某些实施方案中,顺式阻遏序列与报告序列之间间隔1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、35、40、45、50、55、60或更多个间隔核苷酸。7)载体另一方面,本发明的转录本(包括反义和有义序列)自插入dna或rna载体的转录单元而表达(参见例如couture,a,等人,tig.(1996),12:5-10;skillern,a.,等人,国际pct公布号wo00/22113,conrad,国际pct公布号wo00/22114和conrad,美国专利号6,054,299)。这些序列可以作为线性构建体、环状质粒或病毒载体(包括基于噬菌体的载体)的形式来引入,其可作为整合入宿主基因组的转基因而掺入和遗传。转录本也可经构建以允许其作为染色体外的质粒而遗传(gassmann,等人,proc.natl.acad.sci.usa(1995)92:1292)。转录本序列可由位于表达质粒上的启动子来转录。在一个实施方案中,顺式阻遏和报告序列作为由接头多核苷酸序列连接的反向重复序列而表达,使得转录本具有茎和环结构。重组表达载体可用于表达本发明的转录本。重组表达载体通常是dna质粒或病毒载体。表达转录本的病毒载体可以根据但不限于以下病毒而构建:腺伴随病毒(有关综述参见muzyczka,等人,curr.topicsmicro.immunol.(1992)158:97-129));腺病毒(参见例如,berkner,等人,biotechniques(1998)6:616),rosenfeld等人(1991,science252:431-434),和rosenfeld等人(1992),cell68:143-155));或甲病毒以及本领域已知的其它病毒。逆转录病毒已经用于在体外和/或在体内将多种基因引入许多不同的细胞类型,包括上皮细胞(参见例如eglitis,等人,science(1985)230:1395-1398;danos和mulligan,proc.natl.acad.sci.usa(1998)85:6460-6464;wilson等人,1988,proc.natl.acad.sci.usa85:3014-3018;armentano等人,1990,proc.natl.acad.sci.usa87:61416145;huber等人,1991,proc.natl.acad.sci.usa88:8039-8043;ferry等人,1991,proc.natl.acad.sci.usa88:8377-8381;chowdhury等人,1991,science254:1802-1805;vanbeusechem.等人,1992,proc.natl.acad.sci.usa89:7640-19;kay等人,1992,humangenetherapy3:641-647;dai等人,1992,proc.natl.acad.sci.usa89:10892-10895;hwu等人,1993,j.immunol.150:4104-4115;美国专利号4,868,116;美国专利号4,980,286;pct申请wo89/07136;pct申请wo89/02468;pct申请wo89/05345;和pct申请wo92/07573)。可产生能够转导和表达插入细胞基因组的基因的重组逆转录病毒载体,即通过将该重组逆转录病毒基因组转染入合适的包装细胞系,例如pa317和psi-crip(comette等人,1991,humangenetherapy2:5-10;cone等人,1984,proc.natl.acad.sci.usa81:6349)。重组腺病毒载体可用于感染敏感宿主中的各种各样的细胞和组织(例如,大鼠、仓鼠、狗和黑猩猩)(hsu等人,1992,j.infectiousdisease,166:769),并且也具有无需有丝分裂活性细胞用于感染的优势。能够接受待表达的转录本的编码序列的任何病毒载体都可使用,例如,衍生自以下病毒的载体:腺病毒(av);腺伴随病毒(aav);逆转录病毒(例如,慢病毒(lv)、弹状病毒、鼠白血病病毒);疱疹病毒等。病毒载体的向性可以被修饰,即通过用来自其它病毒的包膜蛋白或其它表面抗原对该载体进行假型化(pseudotyping),或通过替代不同的病毒衣壳蛋白,如果合适的话。例如,本发明的特征性的慢病毒载体可以用来自以下病毒的表面蛋白来进行假型化:水泡性口炎病毒(vsv)、狂犬病毒、埃博拉病毒、mokola等。可以制备本发明的特征性的aav载体,以靶向不同的细胞,即通过改造载体,以表达不同的衣壳蛋白血清型。构建表达不同衣壳蛋白血清型的aav载体的技术在本领域技术内;参见例如rabinowitzje等人(2002),jvirol76:791-801,其全部公开内容通过引用结合到本文中。选择适用于本发明的重组病毒载体、将核酸序列插入载体用于表达转录本的方法、和将病毒载体递送到目标细胞的方法,都是在本领域技术内。参见例如,dornburgr(1995),genetherap.2:301-310;eglitisma(1988),biotechniques6:608-614;millerad(1990),humgenetherap.1:5-14;andersonwf(1998),nature392:25-30;和rubinsonda等人,nat.genet.33:401-406,其全部公开内容通过引用结合到本文中。病毒载体可以源自av和aav。用于表达本发明特征性的转录本的合适的av载体、构建重组av载体的方法、和将载体递送入靶细胞的方法,描述于xiah等人(2002),nat.biotech.20:1006-1010。用于表达本发明特征性的转录本的合适的aav载体、构建重组av载体的方法、和将载体递送入靶细胞的方法,描述于samulskir等人(1987),j.virol.61:3096-3101;fisherkj等人(1996),j.virol,70:520-532;samulskir等人(1989),j.virol.63:3822-3826;美国专利号5,252,479;美国专利号5,139,941;国际专利申请号wo94/13788;和国际专利申请号wo93/24641,其全部公开内容通过引用结合到本文中。在本发明的特异性的dna质粒或病毒载体中驱动转录本表达的启动子可以是真核rna聚合酶i(例如,核糖体rna启动子)、rna聚合酶ii(例如,cmv早期启动子或肌动蛋白启动子或u1snrna启动子)或通常地rna聚合酶iii启动子(例如,u6snrna或7skrna启动子)或原核启动子,例如t7启动子,只要表达质粒也编码t7rna聚合酶,其是自t7启动子转录所需的。启动子也可指导转基因表达到胰腺(参见例如来自胰腺的胰岛素调节序列(bucchini等人,1986,proc.natl.acad.sci.usa83:2511-2515))。另外,转录本的表达可以被精确控制,例如通过使用诱导型调节序列和表达系统,例如对某些生理调节剂(例如循环葡萄糖水平、或激素)敏感的调节序列(docherty等人,1994,fasebj.8:20-24)。适于在细胞或哺乳动物中控制转基因表达的这类诱导型表达系统,包括通过以下物质的调节:蜕皮素、雌激素、黄体酮、四环素、二聚化的化学诱导物和异丙基-β-d-1-硫代半乳糖吡喃糖苷(iptg)。根据dsrna转基因的预期用途,本领域技术人员将能够选择合适的调节/启动子序列。通常,如下所述地将能够表达转录本分子的重组载体递送入靶细胞,并在其中持续下去。或者,可以使用病毒载体,其提供转录本分子的瞬时表达。如有必要,可以重复给予这类载体。一旦表达后,转录本与靶rna结合并调节其功能或表达。转录本表达载体的递送可以是系统性的,例如通过静脉内或肌内给予,通过给予到来自患者的外植(ex-planted)的靶细胞,然后重新引入患者体内,或通过允许引入所需靶细胞的任何其它方式。通常将转录本表达dna质粒转染入靶细胞,作为与阳离子脂质载体(例如,oligofectamine)或基于非阳离子脂质的载体(例如,transit-tkotm)的复合物。本发明也涵盖用于dsrna-介导的敲减的多重脂质转染,其在一周或更长时间内靶向单个proc基因或多个proc基因的不同区域。将载体成功引入宿主细胞,可以使用多种已知方式来监测。例如,瞬时转染可以用报告物来发出信号,所述报告物例如荧光标记,例如绿色荧光蛋白(gfp)。使用标记,可以确保对离体细胞的稳定转染,所述标记给转染的细胞提供了对特定环境因素(例如,抗生素和药物)的耐受性,例如潮霉素b耐药性。含有重组dna的载体的递送可以通过非生物或生物系统来进行。包括但不限于脂质体、病毒-样颗粒、衍生自噬菌体或病毒的转导颗粒、和接合。8)转录本测定的报告物在某些实施方案中,所述核酸构建体包括报告序列(例如,报告基因序列)。报告基因编码报告分子,当报告分子在细胞中表达时产生信号。在某些实施方案中,所述报告分子可以是可检测的或可选择的标记。在某些实施方案中,所述报告分子可以是荧光报告分子,例如绿色荧光蛋白(gfp)、黄色荧光蛋白(yfp)、青色荧光蛋白(cfp)、蓝色荧光蛋白(bfp)、或红色荧光蛋白(rfp)。在其它实施方案中,所述报告分子可以是化学发光蛋白。报告分子可以是细菌萤光素酶、真核萤光素酶、荧光蛋白、适于比色检测的酶、适于免疫检测的蛋白、适于免疫检测的肽、或充当适体或表现出酶活性的核酸。可选择的标记也可用作报告物。所述可选择的标记可以是例如抗生素耐药性基因。9)用于转录本报告测定的细胞和靶基因可用于检测的细胞实例包括革兰氏阳性菌和革兰氏阴性菌,例如金黄色葡萄球菌、大肠杆菌、肺炎克雷伯氏菌(k.pneumoniae)等;真菌,例如天蓝色链霉菌(streptomycescoelicolor),和其它真菌细胞,包括来自人类、其它哺乳动物、昆虫、无脊椎动物、或植物的细胞。靶转录本可包括任何内源转录本,无论是编码的还是非编码的。靶转录本可源自真核细胞和原核细胞,包括例如,金黄色葡萄球菌细胞中的meca转录本(指示mrsa)、艰难梭菌中的tcdb转录本(指示产毒性艰难梭菌)、和宫颈上皮细胞中的hpve6/e7转录本(指示宫颈癌)。感染因子例如病毒相关的基因,也可以是靶标,包括hiv、hpv等。靶基因的其它实例包括非编码的rna例如转移rna(trna)和核糖体rna(rrna)、以及rnas例如snornas、micrornas、sirnas、snrnas、exrnas和pirnas和ncrnas。实施例以下是执行本发明的具体实施方案的实施例。给出实施例仅为说明性目的,无意以任何方式限制本发明的范围。已经做出努力以确保所用数字(例如,量、温度等)的准确率,但是,当然应当允许某些实验误差和偏差。本发明的实践将利用本领域技术内的蛋白质化学、生物化学、重组dna技术和药理学的常规方法,除非另有说明。这类技术在文献中有充分解释。参见例如,t.e.creighton,proteins:structuresandmolecularproperties(w.h.freeman和company,1993);a.l.lehninger,biochemistry(worthpublishers,inc.,currentaddition);sambrook,等人,molecularcloning:alaboratorymanual(第2版,1989);methodsinenzymology(s.colowick和n.kaplan编著.,academicpress,inc.);remington'spharmaceuticalsciences,第18版(easton,pennsylvania:mackpublishingcompany,1990);carey和sundbergadvancedorganicchemistry第3版(plenumpress)第a和b卷(1992)。实施例1:沉默突变/互补包装系统以下是基于沉默突变/互补的包装系统的设计和构建的实施例,用于产生非复制型转导颗粒。开发包装系统所用的材料列举如下:细菌菌株:n1706,一种大肠杆菌k-12p1c1-100tn9溶原菌载体:y14439(pbhr1骨架)以下genbank检索号(注意:检索号所指代的序列是到本申请的优先权日为止的数据库中列举的那些)或seqidnos可以用于载体骨架和盒序列:x06758(细菌萤光素酶基因luxab)seqidno:1(天然p1pac-位点)seqidno:3(p1裂解复制子,其含有c1阻遏物-控制的p53启动子、启动子p53反义、repl基因和kila基因的框内缺失)seqidno:4(驱动luxab表达的pblast启动子)n1706(pac):paca突变株的构建:paca突变序列的示例性序列见seqidno:2,显示在以下非正式的序列表中。可以实现突变,即通过经由基因合成而构建突变的序列,然后通过等位基因交换方法,用突变序列替代n1706中的天然序列。gwp10001报告载体的构建:gwp10001载体含有pbhr1复制起点(其表现出广泛的革兰氏阴性活性)、用于卡那霉素和氯霉素的两个可选择的标记、天然噬菌体p1pac-位点序列、操作性连接到组成型blasticillin启动子(pblast)的来自哈氏弧菌(vibrioharveyi)的luxa和luxb基因、和p1裂解复制子(其含有c1阻遏物-控制的p53启动子、启动子p53反义、repl基因和kila基因的框内缺失)。图2显示所得载体(gwp10001,seqidno:11),其可以本领域技术人员已知的多种方式来构建,包括经由pcr从其天然来源获得盒或经由载体的基因合成和装配,经由传统的基于限制酶的克隆或替代技术,例如gibson装配。沉默/互补包装系统:包装系统包括用载体pgwp10001互补的paca突变株n1706(pac)。正如本领域技术人员已知,构建该系统的方式可以通过用载体pgwp10001转化n1706(pac)来完成。可将载体pgwp10001维持在转化的n1706(pac)的培养物中,即通过在50ug/ml卡那霉素存在时培养转化体。携带质粒dna的转导颗粒的产生:经由在42°c的热诱导,携带载体pgwp10001的非复制型转导颗粒可以自n1706(pac)转化体而产生。在42°c孵育导致p1裂解周期的诱导,在裂解周期中,前噬菌体从n1706基因组上切除,产生噬菌体结构元件,并且将裂解复制子所形成的pgwp10001多联体的dna包装入后代噬菌体颗粒,如图1所示。随后收集所得细胞裂解物,其含有非复制型转导颗粒,每个都由携带pgwp10001dna的线性多联体的噬菌体p1颗粒组成。实施例2:缺失/互补包装系统以下是基于缺失/互补的包装系统的设计和构建的实施例,用于产生非复制型转导颗粒。开发包装系统所用的材料列举如下:细菌菌株:rn4220是限制缺陷的金黄色葡萄球菌菌株,其是nctc8325的非溶原化衍生物并且是大肠杆菌dna的有效接受者。它首次在以下文献中得以描述:kreiswirth,b.n.等人,thetoxicshocksyndromeexotoxinstructuralgeneisnotdetectablytransmittedbyaprophage(前噬菌体不可检测地传输中毒性休克综合征外毒素结构基因).nature,1983.305(5936):第709-712页。通过用噬菌体φ80α使rn4220溶原化,得到rn10616。ubeda,c.等人,specificityofstaphylococcalphageandsapidnapackagingasrevealedbyintegraseandterminasemutations(整合酶和末端酶突变所揭示的葡萄球菌噬菌体和sapidna包装的特异性).molecularmicrobiology,2009.72(1):第98-108页。st24源自从rn10616中的溶原化的噬菌体φ80α中缺失小末端酶基因ters。ubeda,c.等人,specificityofstaphylococcalphageandsapidnapackagingasrevealedbyintegraseandterminasemutations(整合酶和末端酶突变所揭示的葡萄球菌噬菌体和sapidna包装的特异性).molecularmicrobiology,2009.72(1):第98-108页。载体:可用作盒的来源质粒的质粒的实例,在本发明的一些实施方案中,描述于charpentier,e.,等人,novelcassette-basedshuttlevectorsystemforgram-positivebacteria(用于革兰氏阳性菌的新的基于盒的穿梭载体系统).appl.environ.microbiol.,2004.70(10):第6076-6085页。以下genbank检索号可以用于盒序列:seqidno:5(金黄色葡萄球菌pt181质粒起点或复制拷贝数变体pt181cop-623repc)m21136(teta(m))seqidno:12(pclpb启动子序列)seqidno:9(φ11小末端酶(ters)基因序列)l09137(ampcole1ori)x06758(luxab)m62650(转录终止)ters缺失:ters敲除株st24的构建可以经由基于等位基因-交换策略而完成,导致框内缺失,去除φ80α小末端酶基因的大部分编码序列。该策略的详情描述于ubeda,c.等人,specificityofstaphylococcalphageandsapidnapackagingasrevealedbyintegraseandterminasemutations(整合酶和末端酶突变所揭示的葡萄球菌噬菌体和sapidna包装的特异性).molecularmicrobiology,2009.72(1):第98-108页。ters敲除株的示例性的序列见seqidno:13,(在以下序列表中显示)。seqidno:13是rn10616基因组序列座,显示φ80αters缺失和互补。载体构建:gw80a0001载体是大肠杆菌/金黄色葡萄球菌穿梭载体。该载体含有金黄色葡萄球菌(pt181cop-623repc)和大肠杆菌(cole1ori)复制起点、用于氨苄青霉素(amp)和四环素(tet(m))耐药性的可选择的标记(分别用于在大肠杆菌和金黄色葡萄球菌中选择)、包括其自身启动子的φ11小末端酶(ters)基因序列、操作性连接到组成型金黄色葡萄球菌pclpb启动子的来自哈氏弧菌的luxa和luxb基因、和转录终止序列(tt)。图4显示所得载体(pgw80a0001,seqidno:14),其可以本领域技术人员已知的多种方式来构建。在一个实例中,tet(m)盒和luxab基因可经由pcr扩增从公众可得的pcn36和pcn58载体(charpentier,e.,等人)而得到。pclpb可以经pcr扩增自金黄色葡萄球菌rn4220而得到,ters可以经pcr扩增自rn10616而得到。可得到载体骨架,即通过从公众可得的载体pcn48中去除ermc基因(charpentier,e.,等人),然后经由适当设计的基于限制酶的克隆,可将最终载体pgw80a0001的多个组件装配成该载体骨架。缺失/互补包装系统:包装系统可包括用载体pgw80a0001互补的ters敲除株st24,以产生菌株gw24。正如本领域技术人员已知,构建该系统的方式可以通过用载体pgw80a0001转化st24来完成。可将载体pgw80a0001维持在转化的st24的培养物中,即通过在5ug/ml四环素存在时培养转化体。携带质粒dna的转导颗粒的产生:经由丝裂霉素c诱导方法,携带载体pgw80a0001的非复制型转导颗粒可以自gw24而产生,所述方法首次在大肠杆菌中得以证明并且目前是从溶原化细菌中得到前噬菌体的标准技术。otsuji,n.等人,inductionofphageformationinthelysogenicescherichiacolik-12bymitomycinc(丝裂霉素c在溶原化大肠杆菌k-12中诱导噬菌体形成).nature,1959.184(4692):第1079-1080页。该前噬菌体诱导方法导致φ80α裂解周期的诱导,其中前噬菌体从gw24基因组上切除,产生噬菌体结构元件,并且将pgw80a0001多联体的dna包装入后代噬菌体颗粒,如图2所示。随后收集所得细胞裂解物,其含有非复制型转导颗粒,每个都由携带pgw80a0001dna的线性多联体的噬菌体φ80α颗粒组成。实施例3:缺乏整合酶的基于sapibov2的包装系统以下是基于sapibov2的包装系统的设计和构建的实施例,用于产生非复制型转导颗粒。开发包装系统所用的材料列举如下:以下材料可用于开发缺乏整合酶的基于sapibov2的包装系统。细菌菌株:rn451是用噬菌体φ11溶原化的金黄色葡萄球菌菌株。jp2131是已经用sapibov2溶原化的rn451。参见maiques,e.等人,roleofstaphylococcalphageandsapiintegraseinintra-andinterspeciessapitransfer(葡萄球菌噬菌体和sapi整合酶在种内和种间的sapi转移中的作用).j.bacteriol.,2007.189(15):第5608-5616页。jp2488是菌株jp2131,其中int基因已从sapibov2中缺失(sapibov2δint)。maiques,e.等人,roleofstaphylococcalphageandsapiintegraseinintra-andinterspeciessapitransfer(葡萄球菌噬菌体和sapi整合酶在种内和种间的sapi转移中的作用).j.bacteriol.,2007.189(15):第5608-5616页。噬菌体:经由丝裂霉素c-诱导方法,噬菌体φ11可得自金黄色葡萄球菌株rn0451,所述方法首次在大肠杆菌中描述并且目前是从溶原化细菌中得到前噬菌体的标准技术。otsuji,n.等人,inductionofphageformationinthelysogenicescherichiacolik-12bymitomycinc(丝裂霉素c在溶原化大肠杆菌k-12中诱导噬菌体形成).nature,1959.184(4692):第1079-1080页。启动子:在本实施例中pclpb可用作启动子。clpb基因启动子是组成型启动子,用于控制int基因的表达。金黄色葡萄球菌clpb(pclpb)基因启动子序列在2004年被首次描述。frees,d.,等人,atpasesarerequiredforstresstolerance,intracellularreplicationandbiofilmformationinstaphylococcusaureus(在金黄色葡萄球菌中,应激耐受性、胞内复制和生物膜形成都需要clpatp酶).molecularmicrobiology,2004.54(5):第1445-1462页。在2004年,它也被首次用于在质粒中控制基因表达。arnaud,m.,a.chastanet,和m.debarbouille,newvectorforefficientallelicreplacementinnaturallynontransformable,low-gc-content,gram-positivebacteria(用于在天然的非可转化的、低gc含量的革兰氏阳性菌中的有效的等位基因替换的新载体).appl.environ.microbiol.,2004.70(11):第6887-6891页。使用2004年描述的引物(同上),启动子可得自金黄色葡萄球菌rn4220。φ11/sapibov2δint共-溶原菌(rn451(φ11sapibov2δint))的产生:菌株jp2488(φ11sapibov2δint)可以通过用φ11使jp2488溶原化而产生。φ11ters缺失(rn451(φ11δterssapibov2δint)):菌株rn451(φ11δterssapibov2δint)可以通过从rn451中缺失φ11ters基因而产生(φ11sapibov2δint),正如以下文献所述:tormo,m.a.等人,staphylococcusaureuspathogenicityislanddnaispackagedinparticlescomposedofphageproteins(将金黄色葡萄球菌毒力岛dna包装入由噬菌体蛋白组成的颗粒).j.bacteriol.,2008.190(7):第2434-2440页。将pclpb-int并入金黄色葡萄球菌基因组(rn451(φ11δterssapibov2δintpclpb-int)):rn451(φ11δterssapibov2δintpclpb-int)可以如下产生:首先经由标准分子生物学技术将pclpb和int融合,然后将pclpb-int融合物插入rn451基因组(φ11δterssapibov2δint),然后选择pclpb-int插入到φ11和sapibov2区之外的克隆。仅携带sapibov2δintpclpb-int多联体的φ11颗粒的产生:经由rn451的丝裂霉素-c诱导可产生仅携带sapibov2δintpclpb-int多联体的φ11颗粒(φ11δterssapibov2δintpclpb-int),正如以下文献所述:otsuji,n.等人,inductionofphageformationinthelysogenicescherichiacolik-12bymitomycinc(丝裂霉素c在溶原化大肠杆菌k-12中诱导噬菌体形成).nature,1959.184(4692):第1079-1080页。细胞裂解物含有非复制型转导颗粒,每个都由携带gi-衍生的dna的线性多联体的噬菌体φ11结构蛋白组成。本领域技术人员将会了解如何使用上述材料和本领域众所周知的分子生物学和遗传技术来构建本发明的nrtps。实施例4:基于ters缺失/互补的sars报告转导颗粒以下是基于诱导物报告物的sars报告系统的实例,其使用基于ters缺失/互补的非复制型转导颗粒。报告基因:细菌萤光素酶(luxab)。luxa和luxb基因来自哈氏弧菌。它们缺乏转录启动子和各自含有其自身的核糖体结合位点。spa基因启动子(pspa):spa基因启动子将用于控制luxab基因的表达。pspa-luxab融合物的构建:可将luxab基因与pspa启动子序列融合,使得luxab基因与pspa启动子操作性连接。luxab-表达报告载体的构建经由标准分子生物学技术可以构建luxab-表达报告载体,即通过将pspa-luxab融合产物并入以下所示的穿梭载体的mcs。大肠杆菌/金黄色葡萄球菌穿梭载体携带金黄色葡萄球菌(pt181cop-623repc)和大肠杆菌(cole1ori)复制起点、氨苄青霉素(amp)和四环素(tet(m))耐药性的基因、处于组成型启动子(pclpb)控制之下的φ11小末端酶(ters)基因、多克隆位点(mcs)、和转录终止序列(tt)。盒序列的genbank检索号:j01764(pt181复制子)m21136(teta(m))尚未得到的检索号(pclpb)af424781区:16526..16966(ters)l09137(ampcole1ori)m62650(tt)经由大肠杆菌top10,可以完成载体的增殖,用于进行体外操纵和用于验证操纵,并且最终修饰的载体随后可被引入金黄色葡萄球菌rn0451δters。经由丝裂霉素c-诱导方法,携带穿梭质粒的转导颗粒可以自rn0451δters转化体而产生,所述方法于1959年首次在大肠杆菌中描述并且目前是从溶原化细菌中得到前噬菌体的标准技术。otsuji,n.,等人,inductionofphageformationinthelysogenicescherichiacolik-12bymitomycinc(丝裂霉素c在溶原化大肠杆菌k-12中诱导噬菌体形成).nature,1959.184(4692):第1079-1080页。随后收集细胞裂解物,其含有非复制型转导颗粒,每个都由携带质粒dna的线性多联体的噬菌体φ11结构蛋白组成,其能够报告靶金黄色葡萄球菌细胞中的sars的存在。实施例5:基于ters缺失/互补的β-内酰胺酶报告转导颗粒以下是基于胞内酶报告物-的β-内酰胺酶报告系统的实施例,其使用基于ters缺失/互补的非复制型转导颗粒。报告基因:海肾萤光素酶(ruc)启动子:启动子可以是pblaz。组成型beta-内酰胺酶启动子可以用于驱动ruc基因的表达。笼蔽的底物:笼蔽的腔肠素-磷酸(coelenterazine-phosphate)如以下文献所述:danielsobek,j.r.,enzymedetectionsystemwithcagedsubstrates(用笼蔽的底物的酶检测系统),2007,zymera,inc.pblaz-ruc融合物的构建:可将ruc基因与pblaz启动子序列融合,使得ruc基因与pblaz启动子操作性连接。ruc-表达报告载体的构建:经由标准分子生物学技术可以构建ruc-表达报告载体,即通过将pblaz-ruc融合产物并入以上部分v,a,3),i)中所述的穿梭载体的mcs。经由大肠杆菌top10,可以完成载体的增殖,用于进行体外操纵和用于验证操纵,并且最终修饰的载体随后可被引入金黄色葡萄球菌rn0451δters。经由丝裂霉素c-诱导方法,携带穿梭质粒的转导颗粒可以自rn0451δters转化体而产生,所述方法于1959年首次在大肠杆菌中描述并且目前是从溶原化细菌中得到前噬菌体的标准技术。otsuji,n.,等人,inductionofphageformationinthelysogenicescherichiacolik-12bymitomycinc(丝裂霉素c在溶原化大肠杆菌k-12中诱导噬菌体形成).nature,1959.184(4692):第1079-1080页。随后收集细胞裂解物,其含有nrtps,每个都由携带质粒dna的线性多联体的噬菌体φ11结构蛋白组成,其能够在φ11宿主范围内的活的金黄色葡萄球菌细胞中表达海肾萤光素酶。实施例6:基于ters缺失/互补的胞内分子报告转导颗粒以下是基于胞内分子报告物-的报告系统的实施例,其使用基于ters缺失/互补的非复制型转导颗粒。启动子:启动子可以是pblaz。组成型β-内酰胺酶启动子可以用于驱动ruc基因的表达。可切换的适体:可切换的适体可以被设计和构建,正如以下文献中所述:samiejaffrey,j.p.,coupledrecognition/detectionsystemforinvivoandinvitrouse(用于体内和体外使用的偶联识别/检测系统),2010,cornelluniversity。荧光团底物:相应的荧光团底物连同上述可切换的适体可以被设计和构建,正如以下文献中所述:samiejaffrey,j.p.,coupledrecognition/detectionsystemforinvivoandinvitrouse(用于体内和体外使用的偶联识别/检测系统),2010,cornelluniversity.pblaz-sa融合物的构建:可将sa基因与pblaz启动子序列融合,使得sa基因与pblaz启动子操作性连接。sa-表达报告载体的构建:经由标准分子生物学技术可以构建sa-表达报告载体,即通过将pblaz-sa融合产物并入以上实施例4中所述的穿梭载体的mcs。经由大肠杆菌top10,可以完成载体的增殖,用于进行体外操纵和用于验证操纵,并且最终修饰的载体随后可被引入金黄色葡萄球菌rn0451δters。经由丝裂霉素c-诱导方法,携带穿梭质粒的转导颗粒可以自rn0451δters转化体而产生,所述方法于1959年首次在大肠杆菌中描述并且目前是从溶原化细菌中得到前噬菌体的标准技术。otsuji,n.等人,inductionofphageformationinthelysogenicescherichiacolik-12bymitomycinc(丝裂霉素c在溶原化大肠杆菌k-12中诱导噬菌体形成).nature,1959.184(4692):第1079-1080页。随后收集细胞裂解物,该裂解物含有非复制型转导颗粒,每个都由携带质粒dna的线性多联体的噬菌体φ11结构蛋白组成,其能够在φ11宿主范围内的活的金黄色葡萄球菌细胞中表达sa。实施例7:基于非复制型转导颗粒的报告系统上述非复制型转导颗粒可用于报告系统,用于经由报告分子(例如luxab)的表达来检测活的细菌的存在。当该转导颗粒将报告载体(例如pgw80a0001)引入转导颗粒的宿主范围内的细胞时,在细胞中被细胞转录机制识别的启动子(例如pclpb)能够驱动该细胞内的报告分子的表达。为了试验非复制型转导颗粒作为报告物的功能性,以便用于检测金黄色葡萄球菌细胞的存在,开发了多种mssa/mrsa报告测定。在一个实施方案中,自金黄色葡萄球菌-特异性噬菌体开发出一种非复制型转导颗粒,并且将处于组成型启动子控制之下的细菌萤光素酶基因luxab并入。当非复制型转导颗粒将报告核酸递送入金黄色葡萄球菌时,组成型启动子表达luxab,适于报告活的金黄色葡萄球菌的存在。另外,将抗生素头孢西丁在加入转导颗粒之前、同时或之后加入到含有金黄色葡萄球菌细胞的样品中。如果细胞对头孢西丁并非表型抗性(即不是mrsa),减少或消除了发光,指示细胞是mssa。然而,如果细胞对头孢西丁具有表型抗性(即是mrsa),可观察到增加的或可检测的发光,指示细胞是mrsa。基于非复制型转导颗粒-的活细胞报告测定功能测定了非复制型转导颗粒作为报告物的功能。在101个临床mrsa分离物中检查了基于噬菌体φ80α-的非复制型转导颗粒的转导宿主范围。进行转导测定,即通过将在改良tsb中培养的各细菌分离物的培养物暴露给含有非复制型转导颗粒的gw24细胞裂解物并在含有四环素的固体培养基上培养所得混合物。在该实例中,非复制型转导颗粒携带四环素可选择的标记。用非复制型转导颗粒转导的细胞预期对四环素具有耐药性。另外,经由发光测定来检查转导,即通过将在液体培养基中的各细菌分离物暴露给含有非复制型转导颗粒的细胞裂解物并在孵育周期后评价所得混合物的细菌萤光素酶发光活性。转导测定显示基于φ80α-的非复制型转导颗粒能够转导所有101个mrsa的临床分离物,而不能转导非金黄色葡萄球菌的葡萄球菌。图21显示了转导测定结果,其中将36种四环素-敏感性mrsa暴露给携带pgw80a0001的转导颗粒,然后点种到含有5ug/ml四环素的培养基板上。结果表明所有36种mrsa菌株因用pgw80a0001转导而生长在含有四环素的培养基上。将mrsa分离物点种到含有四环素培养基上、但不暴露给转导颗粒的对照实验,显示无生长(未显示)。此外,经由对分离的质粒进行测序而证实,自转导的mrsa菌株的质粒分离显示pgw80a0001质粒的恢复。因此转导结果证明了报告质粒的复制起点在所有所测的mrsa分离物上都表现出活性。图22说明了从80种mrsa的临床分离物和28种用转导颗粒转导的甲氧西林敏感性金黄色葡萄球菌(mssa)临床分离物所测定的发光。在实验中,将mrsa和mssa的培养物培养至在600nm处的光密度为0.1,再将100ul在改良tsb中培养的培养物与10ul含有转导颗粒的gw24细胞裂解物混合,随后在37°c进一步孵育4小时,然后测定发光。通过加入10ul1mm癸醛溶液(在表达细菌萤光素酶的细胞内触发发光反应的一种醛)进行发光测定。正如所料,从用金黄色葡萄球菌-特异性非复制型转导颗粒转导的mrsa和mssa中都观察到发光。此外,当将头孢西丁在加入转导颗粒的同时加入细胞培养物中时,从mrsa中观察到发光,但从mssa中未观察到发光。因此证明了转导颗粒用于报告mssa和mrsa的存在的能力。发光结果因此证明了驱动luxab表达的启动子在所有所测的金黄色葡萄球菌分离物上都表现出活性。基于非复制型转导颗粒-的活细胞报告mrsa测定的优化–转导颗粒试剂制剂将非复制型转导颗粒试剂的产生和制剂优化为最终制剂。总之,进行15l规模的发酵,使用tsb培养基,包括gw24的过氧化物诱导。用200ml过夜种子培养物接种15l发酵罐批次(接种率1.3%(v/v))。在o.d.0.8用过氧化氢诱导培养物并在诱导后冷却至25°c,不用ph或do控制。次日早上通过切向流过滤(tff)收获培养上清液,目的是从细胞碎片中澄清噬菌体转导颗粒。将材料随后进一步浓缩并渗滤入无明胶的sm缓冲液并贮存于2-8°c,随后进行最终除菌过滤和贮存。该过程的详细概述如下:正如本领域技术人员已知,可使用多种其它试剂和制剂,以得到制剂。基于非复制型转导颗粒-的活细胞报告mrsa测定的优化–生长培养基制剂对于基于nrtp的活细胞报告mrsa测定,优化生长培养基制剂。为了在基于nrtp的mrsa测定中产生发光,需要将培养基平衡,以便金黄色葡萄球菌生长,并且培养基具有足够的阳离子和添加剂浓度,以有利于nrtp转导。已知在本开发研究之前的用于测定的tsbmod培养基具有沉淀的问题,其会影响培养基的稳定性。生长培养基制剂需要在最终制剂中的稳定性,目标是在室温下1年。方法/程序:用于mrsa测定的细胞制备制备有待测试的测定基础培养基,如表2所述,和准备用于mrsa测定的一组代表性培养基改良见表3。表2:用于生长培养基制剂开发的基础培养基表3:用于生长培养基制剂开发的基础培养基改良按照下表4,向各培养基制备物中,加入nrtp和头孢西丁,制备nrtp培养基试剂:表4:mrsa测定生长培养基/转导颗粒试剂组合30ml培养基终浓度头孢西丁5ug/mlgw24裂解物30xmrsa测定按照以下步骤进行:测定板设置:加入198μl噬菌体培养基试剂和2.0μl各稀释度的细菌0.05od和0.005od,在rpmi中(分别大致相当于20,000和2,000cfu/ml)或2.0ulrpmi作为空白。孵育测定板:在37°c将测定板在定轨振荡器上在~100rpm孵育4小时。准备光度计(moleculardevicesspectramaxl):洗涤试剂按照70%乙醇,接着是di水,然后用底物试剂启动。设置软件为fastkinetic,在10个基线点后以250μl/秒注射50μl底物试剂,并在40点读数,每0.25秒一次。进行测定:让板平衡至室温5分钟后,测试各细菌稀释板。分析通过对所有重复和时间点的空白rlu求平均值并加上3个标准差,确定截止。使用softmaxpro,对于各样品确定最大rlu。确定最大rlu是否大于截止rlu,如果是的话,则样品数据用于比较培养基性能。对于待分析的特定稀释度的菌株,将所有最大rlu值对最大rlu(tsbm1中)(培养基在使用中,直到开发开始)标准化。对于具体的培养基及其改良,对所有mrsa菌株的标准化的rlu值求平均值。对于10种不同的mrsa菌株、2种所测细胞稀释度而言,根据具体培养基的rlu,对两个稀释板的平均值求平均值,最终得到一个数值,代表性能的增加倍数。基于nrtp-的活细胞报告mrsa测定的结果截止rlu的测定:对于各空白重复(4),求出所有时间点(25)的rlu的平均值和标准差。对于各板,求出截止,为平均空白rlu加上3个标准差。相对改进的测定:对于各样品(所有稀释度的空白、mssa和mrsa),从softmaxpro中输出最大rlu并与截止rlu比较。对于噬菌体浓度,如果样品有2个数据点大于截止,则最大rlu值用于分析。将数值通过以下而标准化:通过将具体最大rlu除以其对照条件的最大rlu(在tsbm1-原始培养基中的菌株,在所分析的稀释度时)。在10种mrsa中,对于各培养基条件和各稀释度,对所得比率求平均值,如表5所示。两种稀释度的平均值也示于表中。表5:来自多种生长培养基制剂的mrsa测定结果结论bss2-m56在所测试的多种培养基中平均而言表现出最佳性能。基于hepes缓冲液的培养基的性能优于tris-hcl缓冲的培养基。相比tris-hcl,hepes已知是生物学上有利的缓冲系统。基于b2的基础/肉汤的性能优于基于tsb的肉汤。正如本领域技术人员已知,可使用多种其它试剂和制剂,以得到制剂。经由如上所述的类似实验,开发出其它合适的制剂。其它合适的制剂的实例包括在下表6、7和8中。表6:bsc培养基制剂bsc成分量酪蛋白的酶消化物14.5g酵母提取物35.5g氯化钠35.5gα-d葡萄糖7g总体积1l表7:bsc培养基改良bsc-m64化学名称最终(测定)浓度bgp(mm)60.0hepes(mm)10.0licl(mm)84.0bsc至1l表8:转导颗粒培养基改良转导颗粒制剂(pm4)化学物最终(测定)浓度cacl2(m)0.00667mgcl2(m)0.00335hepes(m)0.01000gw24裂解物储液0.01250叠氮钠(%)0.0006水至1ml基于非复制型转导颗粒-的活细胞报告mrsa测定的优化–底物试剂制剂为了在mrsa测定中产生发光,底物试剂必须包括作为萤光素酶的底物的醛。最先开发的脂族醛制剂(在tsb中的4.2mm三癸醛)不稳定并形成异质乳剂,而非溶液。本实施例概述了底物试剂制剂的开发,其致力于这些问题,目标是在室温或2-8°c达6个月稳定性。该实施例描述了开发底物试剂到最终制剂所采取的步骤。方法/程序使用“模式系统”测试所有筛选和稳定性实验,所述系统包括携带luxab-表达质粒的金黄色葡萄球菌菌株rn4220。典型制备和测试方法如下。使用mrsa测定,测试所有证实实验,以确保当模型系统用于筛选新制剂时,在实际测定中的类似结果。设计用于开发底物试剂制剂的实验以改善以下:分析和结果对各样品的动态反应作图并且线性拟合到各读数点的三次重复的平均值。典型地,结果显示为0.1od模型系统细菌的1:2000稀释度,大致相当于10,000cfu/ml或2,000cfu/测定。对于稳定性实验,分析了相对于参考底物制剂而标准化的最大rlu。在各稳定性时间点,各样品的最大rlu相对于参考底物最大rlu而标准化。标准化的最大rlu相对于时间点作图并且对95%ci的线性回归作图。结论对于产生最终底物试剂制剂,根据参考制剂而调节的关键参数概述于表9。表9:试剂制剂开发结果的概述对底物试剂的改良原因4.2mm三癸醛+tsb原始底物试剂去除tsb降低污染的可能性添加1%吐温20改善溶解度用79.45%0.1m柠檬酸-19.55%0.2m磷酸氢二钠缓冲液调节至ph3改善测定性能直接添加三癸醛到浓缩表面活性剂中改善稳定性增加了将底物试剂通过0.2umpes膜的过滤改善稳定性添加0.05%proclin300改善稳定性添加三乙醇胺改善稳定性将1%吐温20改为0.5%曲拉通x-100改善稳定性,改善溶解度将79.45%0.1m柠檬酸-19.55%0.2m磷酸氢二钠缓冲液改为82%0.1m柠檬酸-18%0.1m柠檬酸钠缓冲液,仍然保持在ph3改善测定性能,去除磷酸缓冲液而减少沉淀的可能性添加100ppm消泡剂y30改善测定性能添加0.5%醋酸维生素e改善稳定性,减少沉淀将主要的三癸醛制造商从alfaaesar改为sigma/omegachem改善测定性能将0.5%醋酸维生素e改为1-2%维生素epeg1000改善测定性能,改善溶解度,改善稳定性对于两种不同贮存温度,制备了两份底物试剂制剂,一份贮存在2-8°c和一份在18-24°c。最终底物试剂制剂贮存在2-8°c。制剂:0.5%曲拉通x-100+4.2mm三癸醛+0.5%醋酸维生素e+100ppm消泡剂y30+0.5%三乙醇胺+82%0.1m柠檬酸+18%0.1m柠檬酸钠@ph3+0.05%proclin300。该制剂在2-8°c1个月后不沉淀并且能够检测到mrsa菌株,如同在第0天一样。最终底物试剂制剂贮存在18-24°c。制剂:0.5%曲拉通x-100+6.3mm三癸醛+100ppm消泡剂y30+0.5%三乙醇胺+82%0.1m柠檬酸+18%0.1m柠檬酸钠@ph3+2%a-生育酚-peg1000琥珀酸+0.05%proclin300。该制剂在18-24°c1个月后不沉淀并且能够检测到mrsa菌株,如同在第0天一样。正如本领域技术人员已知,可使用多种其它试剂和制剂,以得到制剂。基于非复制型转导颗粒-的活细胞报告mrsa测定的分析性能检查了优化的nrtpmrsa测定的分析性能,包括对测定的检测限的分析和当用非靶生物攻击时,对测定的交叉反应性和微生物干扰的分析。a)检测限测定经由确定最低量的mrsa细胞,评价了nrtp测定的检测限,代表能够产生相对光度单位(rlu)信号超出空白样品所测的阈值的多种菌株。mrsa菌株包括sccmeci、ii和iv型,以及携带meca基因变体mecc的mrsa菌株(常规fda-批准的mrsapcr测定未能检测的一个mrsa菌株)。以下关键材料用于临床性能研究:生长培养基试剂:bss-m56底物试剂:最终底物试剂制剂贮存在18-24°c,如上所述。转导颗粒试剂:bss-m56基础,含有10ug/ml(即2x浓度)头孢西丁和转导颗粒试剂,如上所述,以2x浓度。lod研究方案:过夜培养:对于各mrsa菌株和mssa阴性对照菌株,在2mltsb中接种先前在tsa板上培养的菌株的一个菌落。过夜mrsa培养物包括5ug/ml头孢西丁。将所有样品在37°c在振荡培养器中孵育过夜。日间培养:将20ul各过夜培养物移入含有2ml生长培养基试剂的新的培养管。将接种物随后在37°c振荡孵育大约1hr45min,直到od(600nm)达到0.1。系列稀释:细菌载量的计数:将行e的5ul各孔点种在tsa板上,随后将其倾斜,让液滴扩散到板上(为了随后便于菌落计数)。(行e是行a的10-4稀释度)。然后将各板在37°c孵育过夜。测定制备:a)在白色96孔测定板的各孔中填充100ul2x转导颗粒试剂。b)行f和g(即分别是行a的10-5和10-6-倍稀释度)随后用于填充含有转导颗粒试剂的96孔测定板的各孔,使得以一式四份将各样品加入到板上。c)随后用可透气的密封膜(breathableseal)将测定板密封并在37°c孵育4小时,同时中速振荡,50rpm。4小时结束时,将测定板从培养箱中取出并立即在spectramaxl上检测发光,其中注射50μl底物试剂并在1分钟的时间内检测发光。分析:将来自各样品的发光数据作为rlu相对于时间作图。空白样品用于确定根据空白样品的所有时间点而计算的截止,使用以下公式:(平均空白rlu+3*sd空白rlu)随后,对于各样品,得到底物注射后的平均峰值rlu,以确定所产生的rlu值超过空白样品截止的最高稀释度的样品。根据计数研究,确定所产生的rlu值超过空白样品截止的最高稀释度的菌落形成单位(cfu)计数,该cfu计数被报告为研究中的lod。结果:测定了所测的所有mrsa样品的lod都低于10cfu。表11概述了在研究中得到的最低lod的结果。表11:在lod研究中得到的最低lod的结果。sccmec型lod(cfu)i3ii2iv3mecc1所测的所有mrsa菌株都导致小于10cfu,用nrtp测定,高于根据空白样品而计算的截止而检测。mssa不产生高于空白样品截止的rlu值。对于各样品所测的4次重复,rlu值显示在最高稀释度,其中将所产生的高于空白样品截止的rlu值作图,作为平均rlu值和标准差。水平轴设定在空白样品截止,并且产生各rlu数据点的样品的cfu计数用数据叠加。所有mrsa样品产生的rlu值高于截止,而mssa却并非如此。交叉反应性和微生物干扰研究进行了交叉反应性和微生物干扰研究。研究的目的是在mrsa测定中测试在临床样品中常见的并且已知可能在噬菌体φ80α的宿主范围中的一组细菌菌株,观察这些菌株与试验中使用的噬菌体或底物是否有交叉反应性或干扰。用临床样品的先前实验已经导致假阳性结果,当接种在bbl™chromagar™金黄色葡萄球菌板上时,正如蓝色和白色菌落的存在所指示的那样,存在粪肠球菌和表皮葡萄球菌(staphylococcusepidermidis)。另外,单核细胞增多性李斯特氏菌(listeriamonocytogenes)和无害李斯特氏菌(listeriainnocua)可在噬菌体φ80α的感染性或穿透性宿主范围内,其也可有助于在mrsa测定中的交叉反应性。用成活力mrsa测定,本研究测试了粪肠球菌、表皮葡萄球菌、单核细胞增多性李斯特氏菌和无害李斯特氏菌的交叉反应性/干扰。在测定体积中以数量级为106、107或108个细胞的高细胞数量,测试了各菌株。进行测试,没有添加gw24裂解物,以解决可能的菌株自发光。实验1在正常测定条件下,以高细胞数量,测试多种菌株(mssa-s121,nrs#9-溶血性葡萄球菌(staphylococcushaemolyticus)、nrs#6-表皮葡萄球菌、atcc12228-表皮葡萄球菌、atcc15305-腐生葡萄球菌(staphylococcussaprophyticus)、atcc29212-粪肠球菌、atcc60193-白色假丝酵母(candidaalbicans)、atcc12453-奇异变形杆菌(proteusmirabilis))的发光。实验2:在多个浓度的多种抗生素存在时,来自实验1的发光的一个亚组的菌株被重新测定,以猝灭背景发光。实验3:用如上所述开发的多种底物制剂测试粪肠球菌和s32(mrsa),没有gw24裂解物,没有孵育。实验4:测试了atcc33090-无害李斯特氏菌和atcc19111-单核细胞增多性李斯特氏菌的背景信号和非特异性发光,并用如上所述开发的多种底物制剂以及粪肠球菌和表皮葡萄球菌重新测试。实验5:用如上所述开发的最终底物制剂,重新测试粪肠球菌。本研究中测试的底物试剂制剂概述于表9。表10:底物试剂制剂方法/程序:以下是用于进行mrsa测定的步骤。a)菌株培养用于实验1-5在测定前一天,在深96孔板中开始过夜培养,将冷冻的一次性使用的储液在tsb中稀释1:50,并且在37°c在定轨振荡器上孵育>15小时。细菌(8ul)在tsb(392ul)中。在versamax上测定培养物的吸光度。在softmaxpro的模板中将tsb设为空白。在600nm处测定光密度(od)。在测定当天,将细胞重悬至od0.5,以建立测定。制备bss-m56用于实验1-5。b)对于所有实验1、2、4和5,制备转导颗粒培养基试剂(在实验3中未使用转导颗粒试剂):15ug/ml头孢西丁+如上所述的gw24裂解物储液,在30x。c)样品制备:从菌株的过夜培养物制备多个稀释度。所有菌株被稀释bssm56。d)进行mrsa测定,用于实验1-5培养基中装载或不装载噬菌体和头孢西丁5µg/ml到测定板。加入2.5ul细胞。测定板连同板盖一起,在37°c在定轨振荡器上孵育,速度设定在大约100rpm,达4小时。随后,用以下标准测定参数,在spectramaxl上检测各测定板:fastkinetic发光对20个时间点读数,以0.5秒间隔。底物用m注射器注射,50ul/孔,以250ul/秒,包括5个基线读数。未设定孵育温度并在室温下读数。在进行测定前,用底物试剂启动spectramaxl。结果分析如下:a)通过对所有重复和时间点的空白rlu求平均值并加上3个标准差,确定截止。b)使用softmaxpro,对于各样品确定最大rlu。c)确定最大rlu是否大于截止rlu,如果是的话,则样品数据用于分析。结果概述实验1:在mrsa测定中,测试了多种菌株的交叉反应性和干扰,使用原始底物制剂,在所测试的那些中,nrs#9-溶血性葡萄球菌、nrs#6-表皮葡萄球菌和粪肠球菌测试假阳性。实验2:在所测的所有头孢西丁条件下,在所测试的3个菌株中,nrs#9和粪肠球菌测试mrsa阳性。所有3个菌株(nrs#9、粪肠球菌、nrs#6)测试阳性,当测定中未使用转导颗粒试剂时,表明非特异性发光不是转导颗粒试剂-依赖性的,而是菌株和底物试剂依赖性的。所测的所有浓度的carb(羧苄青霉素)都能有效去除假阳性信号。实验3:粪肠球菌给出阳性信号,无转导颗粒试剂。mrsa菌株s32也给出阳性信号,无转导颗粒试剂。该结果指示底物试剂导致背景发光。底物4能有效消除测定中的背景信号。实验4:用转导颗粒试剂和底物试剂,测试了菌株atcc33090-无害李斯特氏菌、atcc19111-单核细胞增多性李斯特氏菌的发光,因为李斯特氏菌(listeriasp)可在mrsa测定中所用的噬菌体的宿主范围内。从无害李斯特氏菌观察到发光,有或无转导颗粒试剂,使用原始底物制剂,表明发光是因为与底物的潜在的非特异性反应。底物5能有效消除李斯特氏菌的发光、而非粪肠球菌的发光。实验5:用底物6重新测试粪肠球菌。在不同的两天进行的两次独立实验中,用0.5od的高载量细胞,测定得到阴性结果。结论交叉反应性研究证明了高载量的若干细菌物种的背景发光。光输出无需转导颗粒试剂和某些底物制剂利用磷酸根离子有助于非特异性信号。因为在使用转导颗粒试剂时,未观察到来自交叉反应性物种的光输出,在这种情况下φ80α穿透交叉反应性物种,因缺乏与细菌萤光素酶基因操作性连接的金黄色葡萄球菌pclpb启动子活性和/或在这些物种内缺乏金黄色葡萄球菌pt181复制起点的活性,而阻止光输出。用柠檬酸钠和柠檬酸替代制剂中的磷酸氢二钠的缓冲液,消除了来自所测的所有交叉反应性物种(除了粪肠球菌之外)的背景发光。底物6和添加的生育酚-peg1000琥珀酸酯的成分消除了来自粪肠球菌的剩余的非特异性信号。基于非复制型转导颗粒的活细胞报告mrsa测定的临床性能-相对于直接接种到chromagarmrsaii上的结果开发了mrsa筛选测定,采用基于φ80α的表达luxab的非复制型转导颗粒(nrtp)。测定包括将nrtp加入疑似含有mrsa的临床样品,将样品在37°c孵育4小时,然后测定孵育的样品,即通过将醛注入样品,同时用光电倍增管检测发光。将测定结果与用作参考的经设计用于检测mrsa的市售的发色培养基进行比较,以确定测定的灵敏性和特异性。基于nrtp的测定预期与基于培养的参考具有良好相关性,因为这两者都需要活的mrsa细胞的存在并且这两者都依赖于mrsa表型的表达。结果显示出与参考的极好相关性。研究的目的是,自测试为mrsa筛选的目的而收集的残余鼻拭子样品,确定基于nrtp的mrsa测定相对于chromagarmrsaii的性能。范围:为了mrsa监测的目的,临床机构自患者收集鼻拭子样品,测试了未鉴定的(de-identified)鼻拭子样品中mrsa的存在,使用基于nrtp的mrsa测定、chromagarmrsaii、chromagarsa和血琼脂tsa,经由直接接种平板和经由富集培养后再接种平板。将基于nrtp的mrsa测定的结果与chromagarmrsaii测定的结果进行比较,以便相对于chromagarmrsaii而计算基于nrtp的mrsa测定的灵敏性和特异性。以下关键材料用于临床性能研究:生长培养基试剂:bss-m56底物试剂:最终底物试剂制剂贮存在18-24°c,如上所述转导颗粒试剂:bss-m56基础,含有10ug/ml(即2x浓度)头孢西丁和转导颗粒试剂,如上所述,以2x浓度。方法/程序临床样品描述:将含有液体amies(220093-bdbbl™培养物拭子™liquidamies)的样品运输管提供给临床机构,用于由临床机构收集未鉴定的残余鼻拭子。在将鼻拭子放入所提供的样品运输管之前,临床机构用拭子进行他们自己的直接培养mrsa筛选,即通过用拭子在培养板上划线。更具体地讲,按照临床机构内部标准程序和使用临床机构的标准收集拭子,收集鼻前孔标本。临床机构再用拭子进行直接培养筛选。再将残余拭子放入样品运输管,其中将拭子头浸入样品运输管中的amies缓冲液中。然后样品在室温保持2-24小时,再进一步处理。样品处理:收到后,将样品贮存在生物安全橱中在室温过夜,竖立放置,以确保拭子浸入样品运输管的amies缓冲液中。过夜贮存后,将样品进一步处理如下。临床样品制备使用1ml移液管,将300µl生长培养基试剂加入15mlfalcon管。从原始运输管中取出来自残余鼻拭子的拭子并浸入相应的falcon管中的生长培养基试剂。然后,通过在生长培养基试剂中前后滚动4-6次,将拭子内容物洗脱入falcon管中的生长培养基试剂。再将拭子放回原始运输管并贮存于2-8°c直到研究结束时,同时将falcon管中的洗脱的临床样品移入1.5ml管并保持在室温,直到进一步处理。进行nrtpmrsa测定:将以下样品直接加载如白色96孔测定板。临床样品:100µl单独(insinglet)的各临床样品的洗脱的材料。mrsa阳性对照:一式三份地将2µl充分混合的0.1od已知mrsa分离物的培养物加入98ul生长培养基试剂。mssa阴性对照:一式三份地将2µl充分混合的0.1od已知mssa分离物的培养物加入98ul生长培养基试剂。空白:100µl生长培养基试剂,一式三份。向各样品中加入100µl转导颗粒试剂。然后将测定板放入设定在37°c的培养箱中,在定轨振荡器上振荡4小时。4小时结束时,将测定板从培养器中取出并立即在spectramaxl上检测发光,其中注射50μl底物试剂并在1分钟的时间内检测发光。将细菌接种平板,用于临床样品cfu计数:经由直接和富集培养,将各洗脱的临床样品铺板,以在chromagarmrsaii、chromagarsa和血琼脂(tsaii)上测定细菌菌落计数,如下。通过直接铺板测定生物体的cfu计数。通过铺板在chromagarmrsaii上测定mrsacfu计数。通过铺板在chromagarsa板上测定金黄色葡萄球菌cfu计数。通过铺板在血琼脂tsa上测定血琼脂tsa支持其生长的任何生物体的cfu计数。在因生物体的载量低于所用平板的检测限而在直接铺板不产生菌落的情况下,也通过将一部分洗脱的临床样品在tsb在37°c振荡孵育过夜,进行样品富集,然后再将富集培养物铺板在chromagarmrsaii上。所有平板都在37°c孵育20-24小时。孵育后,记录各板上出现的任何菌落的cfu计数。分析:根据分别在chromagarmrsaii、chromagarsa和血琼脂tsa上得到的cfu计数,计算每份洗脱的临床样品中的mrsa、金黄色葡萄球菌和总生物体的存在和cfu载量。nrtp测定分析:将来自各样品的数据作为rlu相对于时间作图。截止确定:根据空白样品的所有时间点而计算测定截止,使用以下公式:(平均空白rlu+3*sd空白rlu)。mrsa阳性测定:测定底物注射后的各时间点的rlu高于或低于测定截止。如果注射后的两个或更多个数据点都高于测定截止,则样品被称为“mrsa阳性”。结果:将nrtp测定的mrsa阳性结果与直接和富集培养铺板在chromagarmrsaii上的那些进行比较。进行以下计算以测定ntrp测定的灵敏性和特异性,相对于chromagarmrsaii而言。相对于直接铺板在chromagarmrsaii上的结果表11显示了比较nrtp测定以及直接铺板在chromagarmrsaii上,得到以下结果。表11.nrtp测定结果相比直接铺板在chromagarmrsaii上的结果总样品chromagarmrsaii阳性chromagarmrsaii阴性nrtp测定阳性nrtp测定阴性真阳性真阴性假阳性假阴性69762125775750根据以上数据,相对于直接接种在chromagarmrsaii上,测定的灵敏性和特异性经计算为:灵敏性=100%特异性=92%基于非复制型转导颗粒-的活细胞报告mrsa测定的临床性能-相对于富集培养后铺板在chromagarmrsaii的结果根据相对于直接铺板在chromagarmrsaii的结果,相对于富集培养后铺板在chromagarmrsaii,重新测试所有临床样品。后续测试的基本原理是根据以下可能性:当与直接铺板相比的假阳性结果可能的确是通过nrtp测定检测的真阳性,但可能是直接铺板漏掉了。经由如上所述的nrtp测定,重新测试了一份剩余的洗脱的拭子样品。另一份剩余的洗脱的拭子样品也经由富集培养后铺板chromagarmrsaii而测试。富集培养测试包括将100ul剩余洗脱的拭子材料加入2mltsb并在37℃振荡孵育18-24小时。再将所得培养物在chromagarmrsaii上划线,以测定培养物中的mrsa的存在。表12概述了来自直接铺板和富集后铺板测定的数据–仅显示了在nrtp测定或chromagarmrsaii上产生mrsa阳性结果的样品。表12:nrtp测定结果相比直接铺板和富集培养后铺板在chromagarmrsaii上仅显示了在nrtp测定或chromagarmrsaii上产生mrsa阳性结果的样品。表13显示了比较nrtp测定相对于临床样品富集培养后铺板在chromagarmrsaii上,得到以下结果。表13.nrtp测定结果相比富集培养后铺板在chromagarmrsaii上的结果总样品chromagarmrsaii阳性chromagarmrsaii阴性nrtp测定阳性nrtp测定阴性真阳性真阴性假阳性假阴性6911581257115710根据以上数据,相对于富集培养后铺板在chromagarmrsaii上,测定的灵敏性和特异性经计算为:灵敏性=100%特异性=98.3%实施例8:基于nrtp的测定用于抗微生物敏感性测试-最小抑制浓度与发光输出的相关性在另一实例中,开发了金黄色葡萄球菌头孢西丁敏感性测定,以确定抑制头孢西丁耐药性金黄色葡萄球菌生长所需的头孢西丁的最小抑制浓度。不同于如上所述的区分头孢西丁敏感性与头孢西丁耐药性金黄色葡萄球菌的mrsa头孢西丁耐药性测定,在本实施例中的mrsa头孢西丁敏感性测定描述了一种测定的开发,以测定在头孢西丁存在时抑制金黄色葡萄球菌生长所需的头孢西丁的最小量。以下关键材料用于临床性能研究:生长培养基试剂:bss-m56底物试剂:最终底物试剂制剂贮存在18-24°c,如实施例7所述。转导颗粒试剂:bss-m56基础,含有10ug/ml(即2x浓度)头孢西丁和转导颗粒试剂,如实施例7所述,以2x浓度mic研究方案。过夜培养:对于各mrsa菌株(nrs35和s7)和mssa阴性对照菌株(mssa121),在2mltsb中接种先前在tsa板上培养的菌株的一个菌落。过夜mrsa培养物包括5ug/ml头孢西丁。将所有样品在37°c在振荡培养器中孵育过夜。日间培养:将20ul各过夜培养物移入含有2ml生长培养基试剂的新的培养管。将接种物随后在37°c振荡孵育大约1hr45min,直到od(600nm)达到0.1。经由铺板的mic测定:a)将各日间培养物在含有头孢西丁4、8、16、32、64和128ug/ml的tsa板上划线。b)将各板在37c孵育18小时,测定生长。nrtp测定制备:a)将白色96孔测定板的各孔填充100ul2x转导颗粒试剂。b)对于各日间培养物,随后将5孔填充100ul日间培养物。c)对于各日间培养物,分别将头孢西丁加入一个孔,使得孔中的头孢西丁浓度为4、8、16、32、64和128ug/ml。d)随后用可透气的密封膜将测定板密封并在37°c孵育4小时,同时中速振荡,50rpm。4小时结束时,将测定板从培养箱中取出并立即在spectramaxl上检测发光,其中注射50μl底物试剂并在1分钟的时间内检测发光。分析:将底物试剂添加后来自各样品的最大发光值作图。mssa样品rlu值用于测定截止,使用以下公式来计算:(平均mssarlu+3*sdmssarlu)。结果:图23显示了在4、8、16、32、64和128ug/ml头孢西丁中的金黄色葡萄球菌生长结果。图24显示了在4、8、16、32、64和128ug/ml头孢西丁存在时,通过nrtp测定得到的rlu值。图24中的x轴设定在mssarlu截止值。如图23所示,mrsanrs25表现出mic为128ug/ml头孢西丁,而mrsas7表现出mic为64ug/ml头孢西丁。相应地,mrsanrs25表现出可评价的发光高于mssarlu截止,对于头孢西丁浓度至多64ug/ml头孢西丁;而mrsas7表现出发光高于mssarlu截止,对于头孢西丁浓度至多32ug/ml。根据以上数据,nrtp测定证明了得自测定的rlu值与mic结果相关并因此nrtp测定可以用于开发抗生素敏感性测定。实施例9:转录本报告测定:通过mrsa的meca基因转录本活化的luxab翻译的rbs-阻断顺式阻遏所致的构象变化的机制如上所述,可以设计报告转录本,使得报告基因序列的翻译被报告基因的核糖体-结合位点(rbs)的顺式阻遏所阻断。以下工具可以用于设计本发明的报告转录本。1)使用二级结构程序来计算rna二级结构,所述程序例如mfold(http://mfold.rna.albany.edu/q=mfold/rna-folding-form)。2)使用软件程序来计算分子间的rna相互作用,所述程序例如rna-rnainteractionprediction,使用integerprogramming(ractip)(http://rna.naist.jp/ractip/)。3)使用visualizationappletforrna(varna)(http://varna.lri.fr/)使rna二级结构可视化。图25显示了基于最低能量构象而产生的meca转录本的二级结构,所述最低能量构象是通过mfold而计算并用varna可视化。末端环23(t23)含有yunr序列uugg,其包括meca转录本序列的碱基1,487-1,490。对meca基因转录本的二级结构的分析揭示了若干ssrna区适于设计顺式阻遏的luxab报告物,其可经由报告物和ssrna区之间的相互作用而脱阻遏。如图26中详述,meca转录本的末端环23(t23)含有yunr共有序列。yunr(嘧啶-尿嘧啶-核苷酸-嘌呤)共有序列已经显示出是天然系统中的分子间的rna复合物的关键靶标。顺式阻遏序列经设计而形成茎-环结构,其具有报告序列的rbs,使得顺式阻遏序列阻断了rna聚合酶与报告序列的rbs的结合。当顺式阻遏茎-环结构的环与meca转录本的t23结合时,报告序列被暴露。如图27所示,将顺式阻遏序列2701添加到luxab基因的5’端和经设计而形成茎-环结构,其阻断了luxa基因的rbs序列(“aaggaa”)2702。顺式阻遏茎-环结构经预测能阻断luxarbs(“aaggaa”)序列,根据在luxab转录本5’端包含顺式阻遏序列的luxab转录本的最低能量构象,正如通过mfold而计算并用varna可视化的那样。顺式阻遏的luxab基因的头61个核苷酸见图7,直到luxa基因的起始密码子aug。rbs序列“aaggaa”包括碱基47-52。报告转录本的这一末端环经设计而与meca转录本的末端环23(t23)相互作用(与之结合),该环含有yunr序列。顺式阻遏序列的末端环经设计而与meca转录本的t23相互作用,使得经由顺式阻遏茎-环结构的环与meca转录本结果的t23的相互作用,顺式阻遏的luxab转录本和meca转录本的杂交暴露了luxa基因的rbs。图28显示了mecat23序列和luxab转录本上的顺式阻遏序列之间的预测的分子间的相互作用,其通过ractip而计算并用varna可视化。线表示meca转录本和顺式阻遏的luxab转录本之间的碱基配对。两个序列之间的相互作用导致luxarbs序列aaggaa的暴露并因此使luxab报告物脱阻遏。实施例10:转录本报告测定:检测靶转录本或基因的方法,使用meca–luxab报告系统在另一个实例中,提供了检测靶meca基因的方法,使用meca-luxab报告系统。在此,meca是靶转录本,luxab是报告分子。1.报告构建体的构建经由标准分子生物学技术可以构建包含编码luxab的报告构建体的载体,即通过将报告构建体并入穿梭载体,所述载体在大肠杆菌和金黄色葡萄球菌中都能增殖。所述载体可含有在大肠杆菌中有功能的复制起点和可选择的标记,其在大肠杆菌中表达并且适于允许在选择性条件下生长且用所述载体转化的大肠杆菌细胞的生长。载体也可含有金黄色葡萄球菌中有功能的复制起点和可选择的标记,其在金黄色葡萄球菌中表达并且适于允许在选择性条件下生长且用所述载体转化的大肠杆菌细胞的生长。经由大肠杆菌的合适的实验室克隆菌株,可以完成载体的增殖,用于进行体外操纵和用于验证操纵,并且最终修饰的载体随后可被引入金黄色葡萄球菌菌株。报告构建体可以被首先引入金黄色葡萄球菌细胞,用于转录构建体和产生报告转录本。2.顺式阻遏的报告转录本的构建提供了构建顺式阻遏的报告转录本的方法,所述报告转录本可结合到meca-靶转录本。可经由标准分子生物学技术构建报告转录本。luxa和luxb基因用作报告基因和可以来自哈氏弧菌。所述基因缺乏转录启动子,且各自含有其自身的核糖体结合位点(rbs)。当luxa和luxb基因在细胞中同时被翻译时,luxa和luxb蛋白复合物形成活性萤光素酶(luxab)。参见farinha,m.a.和a.m.kropinski,constructionofbroad-host-rangeplasmidvectorsforeasyvisibleselectionandanalysisofpromoters(宽宿主范围质粒载体的构建,便于启动子的可见选择和分析).j.bacteriol.,1990.172(6):第3496-3499页。顺式阻遏序列可以位于luxab基因上游和启动子下游并且包括与luxarbs互补的序列。接头序列可分开顺式阻遏序列的互补区和luxa序列。载体转录后,顺式阻遏序列的互补区和luxarbs序列复合,产生茎环,其阻止核糖体的泊靠并因此阻止翻译。报告转录本的茎环经设计而不稳定并形成开放复合物,当它与天然存在的meca转录本序列(是细胞内源性的)相互作用时。为了活化luxa基因序列的翻译,天然meca转录本起到反式活化rna的作用,其结合到顺式阻遏的报告转录本并打开螯合luxa基因的rbs的抑制性茎环。一旦rbs不被顺式阻遏序列螯合,就可发生luxa的翻译。经由将报告序列与顺式阻遏序列上游的组成型启动子操作性连接,完成报告构建体的转录。靶meca基因序列的实例见图29。所述序列是meca基因座dna序列(来自金黄色葡萄球菌金黄色亚种sa40,完整基因组genbank:cp003604.1;seqidno:15)和可以用于产生报告构建体,其包含报告序列和顺式阻遏序列。显示了-10位置2901、转录起始位置2902、rbs2903、编码区(灰色904)和转录终止序列2905。图30显示了根据本发明的实施方案,示例性的meca转录本序列可用于设计报告转录本(seqidno:16)。显示了meca的rbs3001和编码序列3002。图31是根据本发明的实施方案,luxab基因座dna序列的实例,其可用于设计报告转录本。luxab基因座dna序列得自费氏弧菌(vibriofischeri)的萤光素酶α和β亚基的基因luxa和luxb(genbank:x06758.1)(seqidno:17)。显示了-10位置3101、转录起始位置3102、luxa的rbs3103、luxa编码区3104(灰色阴影)、luxb的rbs3105和luxb编码序列(灰色阴影)3106。图32是luxab转录本序列的实例,其可用于设计报告转录本(seqidno:18)。显示了luxa的rbs3201、luxa编码序列3202(灰色阴影)、luxb的rbs3203、和luxb编码序列(灰色阴影)3204。图33是luxab顺式阻遏的转录本序列的实例,其可用于报告转录本(seqidno:19)。显示了顺式阻遏序列(虚线框)3301、luxa的rbs3302、luxa编码序列3303(灰色阴影)、luxb的rbs3304和luxb编码序列(灰色阴影)3305。3.使用报告转录本,检测meca靶转录本存在或不存在的方法提供了实例,使用本发明的报告转录本,用于检测细胞中的meca靶转录本存在或不存在。图34显示了包含载体3400的细胞的实例,所述载体编码报告转录本1410,其中在细胞3401中没有内源meca转录本(例如,细胞的基因组不含meca基因)。在这种情况下,顺式阻遏序列3420结合到luxab基因的rbs3430。在某些实施方案中,顺式阻遏序列3420可以结合到luxa基因的rbs的一部分或所有、luxb基因的rbs、或这两者。该结合事件阻断和阻止luxab基因的翻译,并且在细胞中不产生报告分子(例如,萤光素酶)。因此,未检测到信号,指示在细胞中meca基因不存在。在另一实例中,细胞包括内源meca转录本(例如,细胞的基因组含有meca基因)。图35显示了将载体3400引入细胞3401。载体3401编码报告转录本3410,其包括顺式阻遏序列3420和报告序列(luxa和luxb基因)。当存在于细胞中的meca转录本3510结合到顺式阻遏序列1420时,抑制性发夹环打开和luxa基因的rbs3430被暴露。报告序列(luxa和luxb)的翻译可以发生,导致形成luxab酶3520。luxab酶3520产生可检测的发光信号3530。以此方式,转录本报告载体3400报告了细胞3401中的内源meca转录本3510的存在。尽管根据优选实施方案和多个替代实施方案,已经具体地显示和描述了本发明,但是相关领域技术人员将会知道,在形式和细节上可以进行各种改变而不偏离本发明的精神和范围。本说明书中引用的所有参考文献、公布的专利和专利申请都通过引用全部结合到本文中,用于所有目的。所引用的参考文献1.michaelg.schmidt,d.a.s.,carolinewestwater,josephw.dolan,briand.hoel,philipa.werner,jamess.norris,lauram.kasman,nucleicaciddeliveryandexpression(核酸递送和表达),2005.2.kreiswirth,b.n.等人,thetoxicshocksyndromeexotoxinstructuralgeneisnotdetectablytransmittedbyaprophage(前噬菌体不可检测地传输中毒性休克综合征外毒素结构基因).nature,1983.305(5936):第709-712页.3.ubeda,c.等人,specificityofstaphylococcalphageandsapidnapackagingasrevealedbyintegraseandterminasemutations(整合酶和末端酶突变所揭示的葡萄球菌噬菌体和sapidna包装的特异性).molecularmicrobiology,2009.72(1):第98-108页.4.otsuji,n.等人,inductionofphageformationinthelysogenicescherichiacolik-12bymitomycinc(丝裂霉素c在溶原化大肠杆菌k-12中诱导噬菌体形成).nature,1959.184(4692):第1079-1080页.5.brantl,s.(2007)regulatorymechanismsemployedbycis-encodedantisensernas(顺式编码的反义rna使用的调节机制).curr.opin.microbiol.10,102–109.6.isaacs,f.j.等人(2004)engineeredriboregulatorsenablepost-transcriptionalcontrolofgeneexpression(工程化核糖核酸调节子使得转录后的基因表达调节成为可能).nat.biotechnol.22,841–847.7.pfeiffer,v.等人(2009)codingsequencetargetingbymiccrnarevealsbacterialmrnasilencingdownstreamoftranslationalinitiation(miccrna靶向的编码序列揭示了细菌mrna沉默翻译起始下游).nat.struct.mol.biol.16,840–846.8.opdyke,j.a.等人(2004)gady,asmall-rnaregulatorofacidresponsegenesinescherichiacoli(在大肠杆菌中的酸反应基因的小rna调节剂).j.bacteriol.186,6698–6705.9.carriere,c.,等人,conditionallyreplicatingluciferasereporterphages:improvedsensitivityforrapiddetectionandassessmentofdrugsusceptibilityofmycobacteriumtuberculosis(条件复制型萤光素酶报告噬菌体:提高快速检测的灵敏性并评价结核分枝杆菌的药敏性).journalofclinicalmicrobiology,1997.35(12):第3232-3239页.10.merten,o.-w.andm.al-rubeai,viralvectorsforgenetherapy:methodsandprotocols(基因治疗的病毒载体:方法和方案).methodsinmolecularbiology.第737卷.2011.11.lofdahl,s.,j.e.sjostrom,andl.philipson,cloningofrestrictionfragmentsofdnafromstaphylococcalbacteriophage-phi-11(来自葡萄球菌噬菌体-phi-11的dna的限制片段的克隆).journalofvirology,1981.37(2):第795-801页.12.charpentier,e.,等人,novelcassette-basedshuttlevectorsystemforgram-positivebacteria(用于革兰氏阳性菌的新的基于盒的穿梭载体系统).appl.environ.microbiol.,2004.70(10):第6076-6085页.13.novick,r.p.,i.edelman,ands.lofdahl,smallstaphylococcus-auerusplasmidsaretransducedaslinearmultimersthatareformedandresolvedbyreplicativeprocesses(小的金黄色葡萄球菌质粒作为通过复制过程形成和分解的线性多聚体转导).journalofmolecularbiology,1986.192(2):第209-220页.14.westwater,c.,等人,developmentofap1phagemidsystemforthedeliveryofdnaintogram-negativebacteria(用于将dna递送入革兰氏阴性菌的p1噬菌粒系统的开发).microbiology,2002.148(4):第943-950页.15.norris,j.u.,等人,tissue-specificandpathogen-specifictoxicagentsandribozymes(组织特异性的和病原体特异性的毒性剂和核酶).1999.16.maiques,e.,等人,roleofstaphylococcalphageandsapiintegraseinintra-andinterspeciessapitransfer(葡萄球菌噬菌体和sapi整合酶在种内和种间sapi转移中的作用).j.bacteriol.,2007.189(15):第5608-5616页.17.frees,d.,等人,clpatpasesarerequiredforstresstolerance,intracellularreplicationandbiofilmformationinstaphylococcusaureus(在金黄色葡萄球菌中,应激耐受性、胞内复制和生物膜形成都需要clpatp酶).molecularmicrobiology,2004.54(5):第1445-1462页.18.arnaud,m.,a.chastanet,andm.debarbouille,newvectorforefficientallelicreplacementinnaturallynontransformable,low-gc-content,gram-positivebacteria(用于在天然的非可转化的、低gc含量的革兰氏阳性菌中的有效的等位基因替换的新载体).appl.environ.microbiol.,2004.70(11):第6887-6891页.19.tormo,m.a.,等人,staphylococcusaureuspathogenicityislanddnaispackagedinparticlescomposedofphageproteins(将金黄色葡萄球菌毒力岛dna包装入由噬菌体蛋白组成的颗粒).j.bacteriol.,2008.190(7):第2434-2440页.20.arthur,m.,等人,thevanssensornegativelycontrolsvanr-mediatedtranscriptionalactivationofglycopeptideresistancegenesoftn1546andrelatedelementsintheabsenceofinduction(在诱导不存在时,vans传感器负面地控制vanr-介导的tn1546和相关元件的糖肽抗性基因的转录活化).j.bacteriol.,1997.179(1):第97-106页.21.karlsson,s.,等人,expressionofclostridiumdifficiletoxinsaandbandtheirsigmafactortcddiscontrolledbytemperature(艰难梭菌毒素a和b及其σ因子tcdd的表达受到温度的控制).infect.immun.,2003.71(4):第1784-1793页.22.danielsobek,j.r.,enzymedetectionsystemwithcagedsubstrates(用笼蔽的底物的酶检测系统),2007,zymera,inc.23.samiejaffrey,j.p.,coupledrecognition/detectionsystemforinvivoandinvitrouse(用于体内和体外使用的偶联识别/检测系统),2010,cornelluniversity.24.good,l.,translationrepressionbyantisensesequences.cellularandmolecularlifesciences(反义序列所致的翻译阻遏),2003.60(5):第854-861页.25.sabine,b.,antisense-rnaregulationandrnainterference(反义-rna调节和rna干扰).biochimicaetbiophysicaacta(bba)-genestructureandexpression,2002.1575(1-3):第15-25页.非正式的序列表。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1