加速剂组合物的制作方法
本发明涉及一种用于快速固化环氧树脂组合物的新颖加速剂以及此类加速剂用于如可用于制造经固化的热固性制品的可固化环氧树脂组合物的可热固树脂组合物的使用。
背景技术:
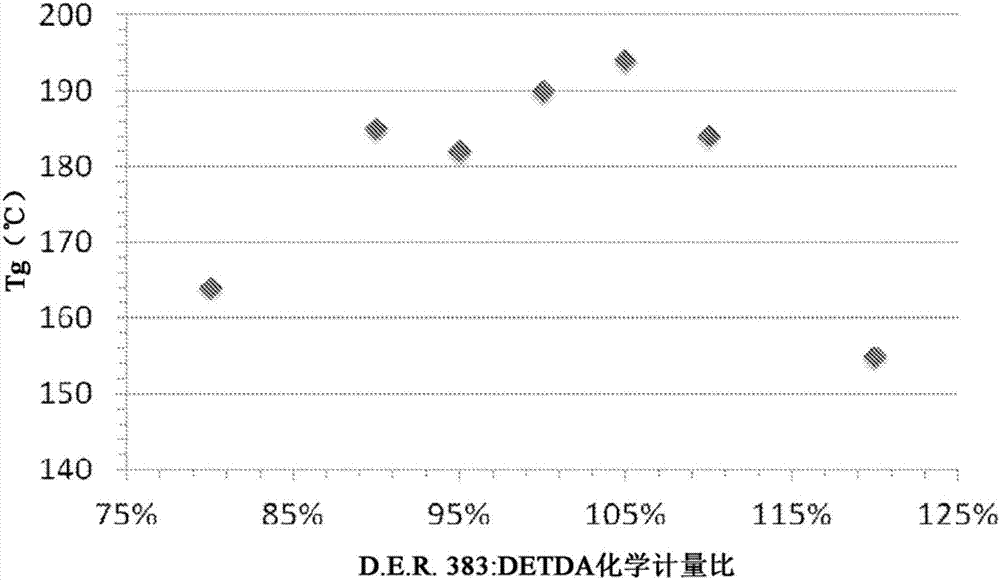
:用于如环氧树脂组合物的可热固树脂的“加速剂”或“催化剂”被定义为催化环氧化物与硬化剂的固化反应的一种化合物或化合物的混合物,并且更精确地,此类“加速剂”加速反应并使反应可以在较低温度下进行或与比不含有加速剂的组合物相比进行较短的时间。一般来说,胺的反应性按以下顺序降低:脂族胺、环脂族胺和芳香族胺。一直存在对加速热固性树脂和胺固化剂之间的固化反应的需要;然而,在当最慢的固化胺,即,芳香族胺与环氧树脂组合物一起使用时的情况下,该需要最为迫切。由于环氧树脂和芳香族胺硬化剂之间的低反应性,所以一般需要较高固化温度和较长固化时间来制造高性能交联的聚合物材料。增大固化温度和固化时间、增加处理成本限制了芳香族胺硬化剂在一些重要复合物制造技术如拉挤成形、树脂传递模制(rtm)、预浸和压缩模制中的应用。为了缩短固化时间和/或降低固化温度,许多催化剂或加速剂用于环氧树脂与胺硬化剂的固化,如苯酚、咪唑、氟化硼-胺络合物、四氟硼酸锌、四氟硼酸铜、三苯基亚磷酸酯、p-电子受体和三烷基锍盐。举例来说,美国专利第5,541,000号公开了一种含有p-电子受体的固化加速剂化合物,其相对于无固化加速剂的对应组合物,可使组合物的固化放热峰值温度降低8百分比至16百分比(%)(例如19℃至28℃),如通过差示扫描量热仪(dsc)使用10℃/分钟(℃/min)扫描速率测定。美国专利第4,447,586号公开了一种四氟硼酸铜水溶液,其可加速含有环氧树脂的可固化组合物和受阻芳香族二胺的固化。然而,在以上专利中描述的固化过程在177℃下需要至少一个小时的固化时间。为实现小于一小时的固化时间,本专利公开需要190℃至195℃的固化温度。美国专利第4,554,342号公开了一种含有低亲核性的阴离子的三烷基锍盐,其可用作用于环氧化合物和芳香族胺组合物的加速剂。在以上专利中教导的加速剂在高温下也需要较长的固化时间,如在80℃下两小时的固化时间接着在150℃下两小时的固化时间。美国专利第6,359,147号公开了羧酸铬(iii),其可催化环氧化合物与羧酸、酸酐、酰亚胺、内酯和碳酸酯的聚合;并且此类催化剂可用于生产预聚物、聚合物和热固性材料。然而,以上专利未教导羧酸铬(iii)用于加速环氧化物和胺之间的反应的适用性。以上现有技术参考文献中无一者公开一种含有过渡金属络合物和盐两者的加速剂,其中该盐的阳离子为金属阳离子或以协同组合具有非亲核性阴离子的鎓阳离子。另外,以上现有技术参考文献中无一者公开了一种经设计用于增加环氧化合物和芳香族胺硬化剂的反应性的加速剂,如通过dsc使用10℃/min扫描速率测定,其使放热起始温度降低大于20%,或在150℃至160℃下小于一小时的固化时间。因此,期望提供一种加速剂,其可有利地且容易地加速环氧化合物和胺硬化剂之间,尤其是环氧化合物和芳香族胺硬化剂之间的反应;并且其可用于可固化调配物中使得当固化调配物时,所得经固化的热固性材料保持包括例如玻璃化转变温度(tg)的性能特性和机械特性的平衡。技术实现要素:一个实施例涉及用于快速固化环氧树脂组合物的协同加速剂,其中加速剂组合物包括至少两种组分,即,(i)过渡金属络合物和(ii)盐的组合;其中过渡金属络合物,组分(i),包括(a)过渡金属离子和(b)配体;其中过渡金属离子,组分(a),可为铬(iii)、zn(ii)或钼(iii);并且其中配体,组分(b),可为衍生自有机酸如辛酸或2-乙基-己酸酯的羧酸根离子。举例来说,过渡金属络合物可为辛酸铬(iii)、辛酸锌(ii)或辛酸钼(iii)。盐,加速剂组合物的组分(ii),包括(c)阳离子,其中阳离子包括金属离子或鎓离子;以及(d)阴离子,其中阴离子包括非亲核性阴离子。举例来说,盐的金属阳离子,组分(c),可包括过渡金属如cu、co、zn、cr、fe、ni等;并且盐的鎓离子,组分(c),可为例如烷基铵离子、芳烷基铵离子或芳基铵离子;或烷基鏻离子、芳烷基鏻离子或芳基鏻离子。盐的非亲核性阴离子,组分(d),可为例如bf4-、pf6-、asf6-、sbf6-等。另一实施例涉及一种可固化环氧树脂组合物,其包括(a)至少一种环氧树脂、(b)至少一种硬化剂(例如,其中硬化剂为胺如二乙基甲苯二胺(detda),和(c)至少一种加速剂组合物,其中加速剂组合物包含本发明的以上加速剂组合物。本发明的加速剂在环氧树脂组合物中的使用可引起环氧树脂组合物的极其快速固化,同时保持环氧树脂组合物的其它品质特性如长适用期、在室温下(例如约20℃到约25℃)的良好时延、低毒性、低粘度和优异机械特性。一种用于确定加速剂如何加速环氧化物和硬化剂之间的固化反应的便捷方法包括以5℃/min或10℃/min扫描速率使用dsc并比较加速体系与未加速体系的热流-温度曲线。举例来说,含有固化加速剂的可固化环氧树脂组合物相对于无固化加速剂的对应的组合物的放热峰值温度,可降低超过53℃(187℃到134℃,降低28%)。在优选的加速剂负载量条件下,例如(在硬化剂中5%cr(c8h15o2)3和3%cu(bf4)2),放热峰值温度可降低72℃(187℃到115℃,降低39%)。以上降低放热效应可使用以5℃/min扫描速率的dsc测定。通过存在于本发明的加速剂中的两种组分的组合实现了显著且出人意料的协同效应,两种组分为:(i)过渡金属络合物和(ii)盐,其中盐的阳离子为金属阳离子或带有非亲核性阴离子的鎓阳离子。组分(i)或(ii)中的任一者单独对环氧-胺组合物的反应性不产生显著的加速影响。举例来说,过渡金属络合物单独无法加速环氧-胺反应;并且盐(其中盐的阳离子为金属阳离子或带有非亲核性阴离子的鎓阳离子)单独无法使组合物的放热峰值温度降低大于53℃(28%)。包括环氧树脂化合物和胺固化剂;和本发明的以上加速剂的可固化组合物可对可固化组合物提供显著的加速影响。使用以上加速剂的固化过程的显著加速是由于存在于加速剂组合物中的两种组分(例如,过渡金属络合物与这样的盐组合,其中盐的阳离子为金属阳离子或带有非亲核性阴离子的鎓阳离子)的协同效应。带有以上组分组合的加速剂表现为不同于已知的加速剂。并且,相对于无加速剂的对应的树脂组合物,含有以上加速剂的可固化树脂组合物的放热峰值温度可降低,例如在一个实施例中大于约23℃,并且例如在另一个实施例中大于约53℃。可固化树脂组合物的放热峰值温度降低大于约23℃的现象先前在其它已知的加速剂的情况下尚未实现或报告。虽然仍未完全理解组分(i)过渡金属络合物(例如羧酸铬(iii))和组分(ii)带有非亲核性阴离子的盐之间的协同效应的机制;但是理论上认为,络合物的过渡金属(铬(iii))提供未占据的配位位点以与环氧基配位从而引发固化过程。盐的阳离子部分也可能参与这个过程。然后非亲核性阴离子使产生的中间产物稳定并促进环氧化物胺反应。加速剂与上述组分(i)和(ii)一起可超过任一组分单独加速环氧固化反应。在实施例中,加速剂含有可催化环氧-胺组合的反应的(i)过渡金属络合物(例如,羧酸铬(iii)、羧酸锌(ii)、羧酸钼(iii),如辛酸铬(iii)或辛酸锌(ii)或辛酸钼(iii))和(ii)盐,其中盐的阳离子为金属阳离子或带有非亲核性阴离子的鎓阳离子(例如,zn(bf4)2或cu(bf4)2),其中所述胺可包括例如芳基脂族胺、脂族胺、杂环胺、环脂族胺、聚醚胺、芳香族胺及其组合。另一实施例涉及一种用于制备以上加速剂组合物的方法。又一个实施例涉及一种用于制备以上可固化组合物或调配物的方法。以及其它实施例涉及一种由以上可固化组合物或调配物和用于其的方法制成的经固化的热固性产物,如复合物。附图说明以下附图说明本发明的非限制性实施例。对于以下图,基于5℃/min的扫描速率记录差示扫描量热仪(dsc)的热流-温度曲线。除非另外说明,否则加速剂浓度是指硬化剂中的重量百分比;并且环氧化物胺当量比率为1。图1为示出如通过dsc所测量的在加速剂cu(bf4)2和cr(c8h15o2)3的情况下d.e.r.383/二乙基甲苯二胺(detda)的放热峰值的图解说明。在图1中,“3%cr(c8h15o2)3+3%cu(bf4)2”是指在detda硬化剂中3重量%(wt%)的cr(c8h15o2)3和3%cu(bf4)2。图2为示出通过1%cu(bf4)2和1%cr(c8h15o2)3催化的具有各种化学计量比的d.e.r.383/detda的经固化的聚合物的tg的示意性图。(tg通过dsc分析)。图3为示出在加速剂1%cu(bf4)2和1%cr(c8h15o2)3的情况下d.e.r.383/detda的动态机械热分析(dmta)的图解说明。图4为示出如通过dsc所测量的在加速剂1%zn(bf4)2和1%cr(c8h15o2)3的情况下d.e.r.383/detda的放热波峰的图解说明。图5为示出如通过dsc所测量的在加速剂1%cu(bf4)2和1%cr(c8h15o2)3的情况下d.e.r.431/detda的放热峰值的图解说明。图6为示出如通过dsc所测量的在加速剂1%zn(bf4)2和1%cr(c8h15o2)3的情况下d.e.r.383/z的放热峰值和tg的图解说明。具体实施方式实施例涉及提供一种加速剂,所述加速剂可显著加速环氧化合物和胺硬化剂之间的反应同时仍维持所得热固性材料的优异机械特性和化学特性。举例来说,实施例涉及提供一种使用芳香族胺硬化剂(例如detda)的环氧树脂组合物以能够实现具有良好耐化学性的高tg交联聚合物的形成。使用芳香族胺硬化剂的交联聚合物一般可用于多种复合物处理技术中,如油井铸造、地热管道、化学管道和槽应用、长丝卷绕、树脂传递模制、预浸、压缩模制等。胺硬化剂还呈现多种有利的特性,如长适用期、良好时延、低毒性、低粘度和对湿气不敏感性。术语在本文中“快速固化”,参考固化组合物,意指环氧化物与硬化剂的固化反应可在至多160℃的固化温度下进行较短时间(例如,几分钟到小于约60分钟)。如根据dsc扫描测定的“起始温度”在本文中意指放热峰的切线与外推基线的相交点。在本文中“对环氧-芳香族胺组合物的反应性的显著加速影响”,参考固化组合物,意指显著降低用于固化环氧化物与硬化剂的反应的温度或时间。举例来说,在下文所述的实例2。中,固化反应的放热峰值温度可降低72℃(187℃到115℃,降低39%),并且固化反应的起始温度可降低54℃(144℃到90℃,降低37.5%)。在本文中“适用期”意指对于较低粘度(例如<1000mpa-s)产品,组合物的初始粘度达到两倍或四倍所花费的时间量。计时从混合组合物的组分的时刻开始,并且在室温下测量粘度。在本文中“长适用期”,参考组合物,意指包括环氧树脂、硬化剂和加速剂的调配物的粘度缓慢增大。举例来说,在下文所述的实例2中,发现d.e.r.383/detda/cu(bf4)2和cr(c8h15o2)3的适用期为超过3小时(hr)并且发现胶凝时间为约40hr。在本文中“胶凝时间”,参考固化组合物,意指混合物不能流动,并且在分子中术语胶凝时间是指形成无穷大网状物的时间点。环氧树脂组合物的胶凝时间通过gardner标准模型胶凝计时器(在25℃下混合物的胶凝时间)和gelnorm胶凝计时器(在80℃和100℃下混合物的胶凝时间)测定。在本文中“时延”,参考硬化剂,意指硬化剂示出在室温下与环氧化合物非常低或有限的反应性,但在高温下硬化剂与环氧化合物反应。举例来说,在下文所述的实例2中,在加速剂[cu(bf4)2和cr(c8h15o2)3]的情况下d.e.r.383/detda在25℃下的胶凝时间为约40hr,但在150℃下的胶凝时间仅为59s。这种环氧树脂组合物显示出良好的时延。流体的粘度为流体对藉由剪应力或张应力渐进变形的阻力的量度。在本文中“低粘度”,参考组合物,意指在50℃下组合物的粘度低于至少1000mpa-s的粘度。举例来说,在50℃下,在实例2中示出的在加速剂cu(bf4)2和cr(c8h15o2)3的情况下组合物d.e.r.383/detda的粘度小于约355mpa-s。在本文中“优异机械特性”,参考组合物,意指高拉伸强度、拉伸模量、拉伸应变、断裂韧度等。举例来说,在下文所述的实例1中,环氧化合物/芳香族胺/加速剂的组合物显示出优异的机械特性,其中拉伸模量为2.6gpa、拉伸强度为56mpa并且断裂韧度为0.5mpa·m1/2。在本发明最广泛范围中,本发明包括加速剂组合物,其包含至少两种组分:(i)(a)过渡金属和(b)配体的络合化合物;其中过渡金属可为铬(iii)、锌(ii)和mo(iii)并且配体可为氧供体配体如辛酸盐或乙酰基丙酮酸盐(例如辛酸铬(iii)或乙酰基丙酮酸铬(iii))或锌(ii)过渡金属和辛酸盐配体(例如辛酸zn);以及(ii)盐,其中盐的阳离子,组分(c),为金属阳离子或鎓阳离子,并且其中盐的阴离子,组分(d),为非亲核性阴离子(例如cu)bf4)2或zn(bf4)2)。本发明的新颖加速剂有利地可用作加速环氧树脂和胺之间的固化反应并且用于可固化热固性树脂组合物中。一般来说,加速剂组合物的至少一种过渡金属络合物可为羧酸铬(iii),cr(oocr)3,其中r为2到约20个碳原子的烷基、芳基、烷芳基和芳烷基基团。加速剂含有过渡金属络合物(i),其由过渡金属离子和配体组成。过渡金属离子可为铬(iii)、zn(ii)或钼(iii)。加速剂组合物的配体组分可包括氧供体配体如羧酸盐。在羧酸失去其氢离子之后,从羧酸获得羧酸盐。羧酸包括例如以下化合物中的一种或多种:c2-c20经取代和未经取代的直链和支链烷基、芳基和芳烷基羧酸,如乙酸、丙酸、己酸、庚酸、辛酸或芳香族酸如苯甲酸;及其混合物。氧供体配体还包括β-二羰基类型化合物如乙酰丙酮。当乙酰丙酮失去氢离子时,阴离子形成所谓的乙酰丙酮酸盐,其为过渡金属络合物的配体组分。过渡金属络合物的配体组分还可为氮供体配体如乙二胺、二亚乙基三胺,或混合的氮和氧供体如三乙醇胺等。在一个优选实施例中,可用于本发明的羧酸铬(iii)化合物可包括辛酸铬(iii)、2-乙基己酸铬(iii)、乙酸铬(iii)、庚酸铬(iii)、乙酰丙酮酸铬(iii)、辛酸锌(ii)或2-乙基己酸zn(ii)、2-乙基己酸钼(iii)及其混合物。使用调配的辛酸铬(iii)如可商购的(尺寸技术化学体系(dimensiontechnologychemicalsystems))hycat3000s也是合适的。从制造商已知的是,hycat3000s含有活化的羧酸铬(iii)以及酚和胺组分。一般来说,本发明中使用的过渡金属络合化合物的量可为以树脂-硬化剂-加速剂组合物的总重量计,例如,在一个实施例中0.01wt%到约15wt%;在另一个实施例中约0.05wt%到约10wt%;在再一个实施例中约0.1wt%到约5wt%;以及在又一个实施例中约0.5wt%到约2.5wt%。当过渡金属络合物的浓度小于上述浓度(例如0.01wt%)时,化合物无法加速环氧-胺反应。加速剂含有作为组分(ii)的盐,其中该盐的阳离子为金属阳离子或鎓阳离子并且该盐的阴离子为非亲核性阴离子。金属可为锂、钠、镁、钙、铝、铁、钴、镍、铜、锌、锡、锑、镉、铅、铋并且鎓离子可为烷基、芳烷基或芳基铵或鏻离子。盐的非亲核性阴离子可为例如bf4-、pf6-、asf6-、sbf6-、fecl4-、sncl6-、bicl5-、alf6-、gacl4-、inf4-、tif6-、zrf6-或clo4-。其它合适的阴离子可包括例如(ch3)2(c6h5)2b-、(c6h5)4b-和(c4h7)4b-。可用于本发明的盐可含有或可不含有结晶水。在一个优选实施例中,可用于本发明的盐可包括cu(bf4)2或zn(bf4)2或其混合物。一般来说,可用于本发明的盐的量可为以树脂-硬化剂-加速剂组合物的总重量计,例如,在一个实施例中0.01wt%到约15wt%;在另一个实施例中约0.05wt%到约10wt%;在再一个实施例中约0.1wt%到约5wt%;以及在又一个实施例中约0.5wt%到约2.5wt%。当盐的浓度小于上述浓度(例如<0.01wt%)时,化合物无法加速环氧-胺反应。然而,当化合物的浓度高于上述浓度(例如>15%)时,化合物无法溶解于环氧-胺组合物中。在另一个实施例中,在加速剂中的羧酸铬(iii),组分(i)和在加速剂中的盐,组分(ii)的化学计量比在约0.01:1到约100:1,优选地0.1:10到约25:1,更优选地约1:10到约10:1的范围内。可用于形成加速剂络合物的任选的组分包括在环境温度或室温下对组分(i)和(ii)实质上惰性的任何增溶剂、溶剂或稀释剂。合适的此类溶剂或稀释剂包括例如醇、酯、二醇醚、酮、脂族和芳香族烃、聚亚烷基二醇、聚亚烷基二醇单醚及其组合等。特别合适的此类溶剂或稀释剂包括例如甲醇、乙醇、丙醇、异丙醇、正丁醇、异丁醇、叔丁醇、叔戊醇、丙三醇、丙酮、甲基乙基酮、甲基异丁基酮、丁二醇甲基醚、乙二醇甲基醚、乙二醇乙基醚、乙二醇正丁基醚、乙二醇苯醚、二乙二醇正丁基醚、二乙二醇乙基醚、二乙二醇甲基醚、丙二醇甲基醚、丙二醇乙基醚、丙二醇正丁基醚、丙二醇苯基醚、二丙二醇甲基醚、二丙二醇正丁基醚、三丙二醇甲基醚、聚乙二醇、聚丙二醇、聚(乙二醇)甲基醚及其任何组合等。在本发明中水的使用不是优选的。一般来说,本发明中使用的溶剂或稀释剂化合物的量可为以树脂-硬化剂-加速剂组合物的总重量计,例如,在一个实施例中0wt%到约20wt%,在另一个实施例中约0.01wt%到约10wt%,和在再一个实施例中0.1wt%到约5wt%,以及在又一个实施例中0.1wt%到约2wt%。一般来说,通过掺合过渡金属络合物(组分(i))、盐(组分(ii))其中盐的阳离子为金属阳离子或鎓阳离子并且盐的阴离子为非亲核性阴离子,以及(iii)任选地,视需要和上述的任何其它任选组分来产生本发明的加速剂组合物。用于制备本发明的加速剂的方法包括将加速剂组分共混或混合到硬化剂中或到环氧树脂中或到环氧树脂硬化剂混合物中。优选的方法为将加速剂组分混合到硬化剂中。在混合过程期间,任选的溶剂可用于促进加速剂的组分的溶解,其目的在于获得均匀的调配物或均匀的硬化剂加速剂混合物或均匀的环氧化物加速剂混合物。用于制备本发明的加速剂的方法包括在在一个实施例中在约-10℃到约100℃,在另一个实施例中约10℃到约80℃以及在再一个实施例中约0℃到约60℃的温度下,共混或混合上述过渡金属络合物、上述盐和任何其它任选的组分。举例来说,可在一个实施例中约1psig(6.9kpa)到约150psig(1,034.2kpa);在另一个实施例中约5psig(34.5kpa)到约80psig(551,6kpa);以及在再一个实施例中约10psig(68.9kpa)到约20psig(137.9kpa)的压力下进行该方法。用于产生加速剂组合物的方法的时间一般可为,在一个实施例中约0.01hr到约20hr,在另一个实施例中约0.1hr到约10hr,以及在再一个实施例中1hr到约2hr。本发明的加速剂组合物的制备和/或其任何步骤可为分批或连续方法。用于进行反应的设备包括对本领域的技术人员已知的设备。通过本发明的方法制备加速剂组合物产物为具有协同、出人意料和独特特性的新颖组合物。举例来说,已发现本发明的加速剂可引起环氧-胺的极其快速固化,同时保持其它特性,如长适用期、在室温下良好时延、低毒性、低粘度、耐化学性和优异机械特性。出人意料地,在一个实施例中,本发明的加速剂可使可固化环氧-芳香族胺组合物的放热峰值温度降低72℃(187℃到115℃,降低39%)。如前述,在本发明的一个实施例中加速剂可用于环氧树脂调配物中,所述环氧树脂调配物通常使用常规的固化剂固化以产生经固化的产物或热固性材料。然而,本发明的优选实施例是为了加速环氧树脂和胺固化剂之间的固化反应。举例来说,含有加速剂的可固化环氧树脂组合物可包括:(a)至少一种环氧树脂;(b)至少一种硬化剂或固化剂,如胺;以及(c)本发明的加速剂;以及(d)任选地,任何其它所需添加剂。本发明的可固化调配物包括作为组分(a)的至少一种环氧树脂。本文所用的环氧树脂可为含有至少两个环氧基的单体化合物、低聚化合物或聚合化合物。环氧树脂可为脂族、环脂族、芳香族、环状、杂环的或其混合物。环氧树脂可为饱和或不饱和的。环氧树脂可为经取代或未经取代的。可用于本发明的大量环氧树脂见于lee,h.和neville,k.的《环氧树脂手册(handbookofepoxyresins)》麦格劳希尔图书公司(mcgraw-hillbookcompany),纽约(newyork),1967,第2章,第257至307页;以引用的方式并入本文中。用于在本发明的本文所公开的实施例中使用的环氧树脂可变化并且包括常规和可商购的环氧树脂。本文所用的树脂组合物的环氧树脂组分可包括单独使用的单一环氧树脂化合物或以组合使用的两种或更多种环氧化合物的混合物。环氧树脂,也被称作聚环氧化物,可为每分子具有平均多于一个未反应的环氧化物单元的产品。在选择用于本文所公开的组合物的环氧树脂中,应考虑最终产物的特性,还应考虑可影响树脂组合物加工的粘度和其它特性。用于本发明的组合物中的合适的常规环氧树脂化合物可通过本领域中已知的方法制备,如例如,基于表卤代醇和(1)苯酚或苯酚型化合物、(2)胺或(3)羧酸的反应的反应产物。本文所用的合适的常规环氧树脂还可由不饱和化合物的氧化来制备。举例来说,本文所用的环氧树脂可包括表氯醇与如下项的反应产物:多官能醇、苯酚、双酚、卤化双酚、氢化双酚、酚醛清漆树脂、邻甲酚酚醛清漆、苯酚酚醛清漆、聚二醇、聚亚烷基二醇、环脂烃、羧酸、芳香族胺、氨基苯酚或其组合。环氧化合物的制备描述于例如kirk-othmer的《化学技术百科全书(encyclopediaofchemicaltechnology)》,第3版,第9卷,第267至289页。在一个实施例中,可用于与表卤代醇反应以制备环氧树脂的合适的酚、酚型或多元酚化合物包括例如,每分子具有平均多于一个芳香族羟基的多元酚化合物,如例如,二羟基苯酚;联苯酚;双酚如双酚a、双酚ap(1,1-双(4-羟苯基)-1-苯基乙烷)、双酚f或双酚k;卤化联苯酚如四甲基-四溴联苯酚或四甲基三溴联苯酚;卤化双酚如四溴双酚a或四氯双酚a;烷基化联苯酚如四甲基联苯酚;烷基化双酚;三酚;酚-醛酚醛清漆树脂(即,苯酚和简单醛优选甲醛的反应产物)如酚-甲醛酚醛清漆树脂、经烷基取代的酚-甲醛树脂、酚-羟基苯甲醛树脂、烷基化酚-羟基苯甲醛树脂或甲酚-羟基苯甲醛树脂;卤化酚-醛酚醛清漆树脂;经取代的酚-醛酚醛清漆树脂;酚-烃树脂;经取代的酚-烃树脂;烃-酚树脂;烃-卤化酚树脂;烃-烷基化酚树脂;间苯二酚;儿茶酚;对苯二酚;二环戊二烯-酚树脂;二环戊二烯-经取代的酚树脂;或其组合。在另一个实施例中,可用于与表卤代醇反应以制备环氧树脂的合适的胺包括例如二氨基二苯基甲烷、氨基苯酚、二甲苯二胺、苯胺或其组合。在再一个实施例中,可用于与表卤代醇反应以制备环氧树脂的合适的羧酸包括例如邻苯二甲酸、间苯二甲酸、对苯二甲酸、四氢邻苯二甲酸和/或六氢邻苯二甲酸、内亚甲四氢邻苯二甲酸、间苯二甲酸、甲基六氢邻苯二甲酸或其组合。可用于本发明的环氧树脂的几个非限制性实施例包括例如由表卤代醇和聚乙二醇的反应制备的脂族环氧化物,如三甲基环氧丙烷;二缩水甘油基-1,2-环己烷二甲酸酯或其混合物;双酚a的二缩水甘油醚;双酚f的二缩水甘油醚;间苯二酚二缩水甘油醚;对氨基苯酚的三缩水甘油醚;含卤素(例如氯或溴)的环氧树脂,如四溴双酚a的二缩水甘油醚;环氧化苯酚酚醛清漆;环氧化双酚a酚醛清漆;经噁唑烷酮改性的环氧树脂;环氧基封端的聚恶唑烷酮;及其混合物。用于在本发明的组合物中使用的合适的可商购环氧树脂化合物可为例如,可商购自陶氏化学公司(thedowchemicalcompany)的环氧树脂,如d.e.r.tm300系列、d.e.ntm400系列,d.e.r.tm500系列、d.e.r.tm600系列和d.e.r.tm700系列的环氧树脂。可用于本发明的双酚a类环氧树脂的实例包括可商购的树脂,如可商购自陶氏化学公司的d.e.r.tm300系列和d.e.r.tm600系列。可用于本发明的环氧酚醛清漆树脂的实例包括可商购的树脂,如可商购自陶氏化学公司的d.e.ntm400系列。举例来说,作为本发明的一个说明性实施例,环氧树脂可为液体环氧树脂,如d.e.r.383,双酚a的二缩水甘油醚(dgeba),其环氧当量(eew)为约175到约185,粘度为约9.5pa-s并且密度为约1.16g/cc。可用于环氧树脂组分的其它市售环氧树脂可为d.e.r.330、d.e.r.354、d.e.r.332或其混合物。可用作组分(i)的其它合适的环氧树脂公开于例如美国专利第3,018,262号;第7,163,973号;第6,887,574号;第6,632,893号;第6,242,083号;第7,037,958号;第6,572,971号;第6,153,719号;以及第5,405,688号;pct公开案wo2006/052727;美国专利申请公开案第20060293172号和第20050171237号中,其中的每一个在此以引用的方式并入本文中。适合用于本发明的组合物的环氧树脂和其前体的实例还描述于例如美国专利第5,137,990号和第6,451,898号中,其以引用的方式并入本文中。可用于本发明的组合物中的环氧树脂的优选实例包括每分子具有至少2个环氧基的脂族和芳香族环氧树脂两者。可用于制备可固化环氧树脂调配物的优选的环氧树脂化合物的几个非限制性实例可包括例如但不限于双酚a、双酚f、苯酚酚醛清漆、环脂族物、联苯酚环氧树脂、二乙烯基芳烃二氧化物;多元醇和表氯醇的加成产物;及其混合物。一般来说,用于本发明的环氧树脂化合物的浓度一般可在以树脂组合物中的组分的总重量计,在一个实施例中约1wt%到约99wt%,在另一个实施例中约10wt%到约90wt%,在再一个实施例中约30wt%到约90wt%,以及在又一个实施例中约40wt%到约85wt%的范围内。一般来说,固化剂(也被称作硬化剂或交联剂),组分(b),与环氧树脂,组分(a)共混,并且将上述加速剂添加到共混物中以制备本发明的可固化组合物或调配物。可固化调配物然后可经固化以形成经固化的产品或热固性材料。在优选的实施例中,可用于本发明的固化剂包括芳香族胺、芳基脂族胺、脂族胺、杂环胺、环脂族胺、聚醚胺及其组合。举例来说,芳香族胺硬化剂可包括具有直接连接到芳环结构的碳原子的两个或更多个伯或仲氨基的那些芳香族化合物。可用于可固化组合物的芳香族胺硬化剂的代表性实例包括但不限于二乙基甲苯二胺;4,4'-二氨基二苯基醚;3,3'-二氨基二苯基砜;1,2-苯二胺、1,3-苯二胺和1,4-苯二胺;双(4-氨基苯基)甲烷;双(4-氨基苯基)砜;1,3-二甲苯二胺、1,2-二氨基-3,5-二甲基苯;4,4'-二氨基-3,3'-二甲基联苯;4,4′-亚甲基双(2,6-二甲基苯胺);1,3-双-(间胺基苯氧基)苯;9,9-双(4-氨基苯基)茀、3,3'-二氨基二苯砜;4,4'-二氨基二苯硫化物;1,4-双(对氨基苯氧基)苯、1,4-双(对氨基苯氧基)苯、1,3-丙二醇-双(4-氨基苯甲酸酯);及其混合物。可用于本发明的芳基脂族胺的实例可包括但不限于间二甲苯二胺(mxda);对二甲苯二胺;及其混合物。可用于本发明的脂族胺的实例包括但不限于乙二胺(eda);二亚乙基三胺(deta);三亚乙基四胺(teta);三甲基己二胺(tmda);己二胺(hmda);n-(2-氨基乙基)-1,3-丙二胺(n3-胺);n,n'-1,2-乙二基双-1,3-丙二胺(n4-胺);二亚丙基三胺;及其混合物。可用于本发明的杂环胺的实例包括但不限于n-氨基乙基哌嗪(naep);3,9-双(3-氨丙基)2,4,8,10-四氧杂螺(5,5)十一烷;及其混合物。可用于本发明的环脂族胺的实例包括但不限于1,3-双氨基环己胺(1,3-bac);异佛尔酮二胺(ipd);4,4'-亚甲基双环己胺;双-(对氨基环己基)甲烷(pacm)及其混合物。可用于本发明的聚醚胺的实例包括但不限于其中烷氧基为氧亚乙基、氧亚丙基、氧基-1,2-亚丁基、氧基-1,4-亚丁基或其共聚物的聚醚胺。举例来说,可用于本发明的聚醚胺可包括但不限于4,7-二氧杂癸烷-1,10-二胺;1-丙胺、2,1-乙二基氧基))双(二氨基丙基化二乙二醇)(1922a);聚(氧基(甲基-1,2-乙二基));α-(2-氨基甲基乙基)ω-(2-氨基甲基乙氧基)(d-230、d-400);三乙二醇二胺和低聚物(xtj-504、xtj-512);聚(氧基(甲基-1,2-乙二基))、α,α'-(氧基二-2,1-乙二基)双(ω-(氨基甲基乙氧基))(xtj-511);双(3-氨丙基)聚四氢呋喃350;双(3氨丙基)聚四氢呋喃750;聚(氧基(甲基-1,2-乙二基));a-氢-w-(2-氨基甲基乙氧基)醚与2-乙基-2-(羟基甲基)-1,3-丙二醇(t-403);二氨基丙基二丙二醇;及其混合物。一般来说,用于本发明的可固化调配物的固化剂的量将取决于可固化组合物的最终用途。举例来说,作为一个说明性实施例,固化剂的浓度一般可为以可固化调配物中组分的重量计,在一个实施例中约1wt%到约99wt%,在另一个实施例中约5wt%到约80wt%;以及在再一个实施例中约15wt%到约60wt%。一般来说,在一个优选实施例中,用于本发明的可固化调配物的固化剂的量可为例如约0.7当量/环氧当量到约1.8当量/环氧当量,以及在另一个实施例中约0.9当量/环氧当量到约1.3当量/环氧当量。在制备本发明的可固化树脂调配物中,将上述加速剂组合物作为组分(c)添加到树脂调配物中。加速剂可用于本发明以促进并加速环氧树脂和胺固化剂之间的反应。一般来说,可固化组合物中使用的加速剂的量可为例如,在一个实施例中0.01wt%到约15wt%(以全部组合物的重量计),在另一个实施例中约0.05wt%到约5wt%;在再一个实施例中约0.1wt%到约2wt%;以及在又一个实施例中约0.4wt%到约1wt%。可调节加速剂含量以允许在最终应用中充分处理。可添加到本发明的可固化组合物中的其它任选化合物可包括通常用于本领域技术人员已知的树脂调配物中用以制备可固化组合物和热固性材料的化合物。举例来说,任选的组分可包含可添加到组合物中以增强应用特性(例如表面张力调节剂或流动助剂)、可靠性(例如助粘剂)、反应速率、反应选择性和/或催化剂寿命的化合物。可添加到本发明的可固化组合物中的其它任选化合物或添加剂可包括例如其它助催化剂、脱模剂;进一步降低调配物的粘度的溶剂,可与可固化调配物中的其它成分共混的其它树脂如酚系树脂、加成固化剂、填充剂、颜料、增韧剂、流动调节剂、助粘剂、稀释剂、稳定剂、塑化剂、催化剂减活化剂、阻燃剂及其混合物。一般来说,当用于本发明中时,其它任选组分或添加剂的量可为例如,在一个实施例中0wt%到约70wt%,在另一实施例中约0.01wt%到约40wt%;在再一实施例中约0.1wt%到约10wt%;以及在又一实施例中约0.2wt%到约5wt%。用于制备本发明的可固化调配物的方法包括掺合(a)环氧树脂化合物;(b)固化剂;(c)上述加速剂;以及(d)任选地,视需要和上述的任何其它任选成分。以上组分可按任何次序添加到混合物中。举例来说,本发明的可固化树脂调配物的制备通过在已知的混合设备中共混上述环氧树脂、上述加速剂、上述固化剂及任选地任何其它需要添加剂来实现。以上提及的任选添加剂中的任一者,如增韧剂、流动调节剂或粘合改性剂等,可在混合期间或在混合之前,添加到组合物中以形成调配物。在另一个实施例中,在本发明的可固化树脂调配物的制备期间,上述加速剂(c)可首先与固化剂(b)混合,并且然后与环氧树脂(a)混合接着在任何任选添加剂中混合。可固化调配物的所有化合物通常都在使得能够制备有效可固化环氧树脂调配物的温度下混合和分散,所述调配物具有关于特定应用的所需的特性平衡。举例来说,在所有组分的混合期间温度一般可为,在一个实施例中约-10℃到约150℃,在另一个实施例中约0℃到约80℃,在再一个实施例中约0℃到约60℃,以及在又一个实施例中约0℃到约25℃。较低的混合温度有助于使组合物中环氧化物和固化剂的反应减到最少而使组合物的适用期增到最大。本发明的可固化调配物的制备和/或其任何步骤可为分批或连续方法。方法中所用的混合设备可为本领域的技术人员熟知的任何容器和辅助设备。可固化调配物的一些益处可包括例如低粘度、长适用期、良好时延、高tg和优异的机械特性。举例来说,通过以上方法制备的可固化环氧化合物调配物有利地呈现低粘度,例如在25℃下小于或等于(≤)约2,000mpa-s的粘度。一般来说,可固化调配物在25℃下的粘度可为,在一个实施例中约500mpa-s到约20,000mpa-s,在另一个实施例中约500mpa-s到约10000mpa-s,以及在再一个实施例中约500mpa-s到约5,000mpa-s。另外,举例来说,通过以上方法使用本发明的加速剂制备的可固化调配物有利地呈现长适用期,如在25℃下大于或等于(≥)约40hr。一般来说,可固化调配物的适用期可为,在一个实施例中在25℃下约0.1hr到约60hr,在另一个实施例中在25℃下约1hr到约40hr,以及在再一个实施例中在25℃下约3hr到约20hr。可固化调配物,当固化时,赋予经固化的热固性材料如由可固化调配物制备的纤维强化复合物具有优异可挠性、耐冲击性、耐化学性和其它特性如防潮性,这可归因于本发明的可固化环氧树脂-胺组合物中加速剂的存在的。下文更详细论述经固化的热固性产物的特性。本发明的另一个实施例包括使上文所论述的可固化树脂调配物固化以形成热固性材料或经固化的制品。举例来说,可在预定温度下进行可热固组合物或可固化树脂调配物的固化并且持续足以固化树脂组合物的预定时间段。举例来说,固化调配物的温度一般可为在一个实施例中约30℃到约180℃;在另一个实施例中约80℃到约170℃;以及在再一个实施例中约120℃到约160℃;并且可选择固化时间,在一个实施例中在约10s到约4hr之间,在另一个实施例中在约2分钟(min)到约2hr之间,以及在再一个实施例中在约2min到约1hr之间。少于约10s的时间段,时间可过短而无法确保常规处理条件下的充分反应;并且多于约4hr,时间可过长而不实用或经济。具有作为本发明加速剂(含有(i)过渡金属络合物(例如辛酸铬(iii))和(ii)盐,其中盐的阳离子为金属阳离子或带有非亲核阴离子的鎓阳离子(例如cu(bf4)2或zn(bf4)2)两者的加速剂络合物)的一种组分的本发明的可固化调配物可用于制造用于各种应用的经固化热固性产物。本发明的经固化产物(即,由可固化调配物制备的交联产物)显示出优于由含有常规加速剂的可固化组合物制备的常规经固化热固性材料的若干改善的和有利的性能特性。当加速剂用于可固化组合物或调配物中时,例如,可固化调配物提供所得经固化组合物具有有利的性能特性如较高tg和更好的机械特性。举例来说,本发明的经固化产物可有利地具有高tg。一般来说,本发明的经固化产物一般呈现在一个实施例中在约60℃和约290℃之间,在另一个实施例中在约120℃和约230℃之间,以及在再一个实施例中在约150℃和约200℃之间的玻璃化转变温度。经固化产物的tg可通过下文实例中描述的方法测量。使用含有本发明的加速剂的可固化组合物已发现的另一个益处为由组合物制备的经固化热固性材料的部分热和机械特性对湿气不敏感,即,如果经固化热固性材料经受湿气;或如果例如通过使用含有结晶水的组分将水引入到调配物中,那么经固化热固性材料的热和机械特性不改变。有利的是,一般来说,组合物特性与组合物的含水量无关。在另一个优点为,由于在多种复合物制造方法中可固化组合物为合适的基质,所以组合物可在玻璃或碳纤维(其中纤维被短切或对齐)存在下固化。本发明的加速剂可用作用于环氧树脂-硬化剂可固化组合物或调配物的加速剂以加速环氧树脂和硬化剂之间的反应。继而,含有本发明的加速剂的可固化环氧树脂调配物可用于其中使用常规环氧树脂的热固性材料体系中。举例来说,其中可使用含有本发明加速剂的环氧树脂调配物的应用的一些非限制性实例包括例如复合物、涂料、粘合剂、墨水、封装、铸造以及传统上使用热固性树脂和固化剂的任何其它应用。还可测量和评估由含有本发明的加速剂的可固化环氧树脂调配物制备的经固化产物的有利特性以确定可固化调配物和经固化产物的所需最终用途。在一个优选实施例中,催化的环氧-芳香族胺可用于例如纤维强化复合物制造的应用中,包括湿式敷涂、输注、长丝卷绕、拉挤成形、树脂传递模制(rtm)、预浸工艺、注射模制和压缩模制。用于制造纤维强化复合物的纤维强化物将通常为,例如作为纤维或垫的玻璃,碳纤维和芳纶纤维。实例以下实例和比较实例进一步详细说明本发明,但不应解释为限制其范围。在以下实例中,使用了各种术语和名称,如例如:“detda”表示二乙基甲苯二胺。“dsc”表示差示扫描量热仪。“dmta”表示动态机械热分析。“doe”表示实验设计。d.e.r.383为具有173至183的eew且可商购自陶氏化学公司的环氧树脂。d.e.r.431为具有172至179的eew且可商购自陶氏化学公司的环氧酚醛清漆树脂。z为用作用于环氧树脂的固化剂的芳香族胺(1,3-苯二胺和4,4'-二氨基二苯基甲烷)的共混物且可商购自空气产品(airproducts)。d230为聚醚胺且可商购自亨茨曼公司(huntsmancorporation)。cu(bf4)2为四氟硼酸铜(ii)水合物且可商购自西格玛-奥德里奇化学公司(sigma-aldrichchemical)。zn(bf4)2为四氟硼酸锌(ii)水合物且可商购自西格玛-奥德里奇化学公司。cr(acac)3为乙酰基丙酮酸铬(iii)且可商购自西格玛-奥德里奇化学公司。cr(c8h15o2)3为2-乙基己酸铬(iii)且可商购自阿法埃莎(alfaaesar)、谢普德化学公司(theshepherdchemicalcompany)或尺寸技术化学体系。在以下实例中,使用标准分析设备和方法,如例如:差示扫描量热法测量使用来自ta仪器(tainstruments)的配备有自动采样器和制冷冷却器系统(rsc)的q2000型号dsc进行差示扫描热量测定(dsc)分析。通过使用富拉克泰克(flacktek)混合器(以2,200转/分钟[rpm]持续2min)混合约10克(g)的调配样品。然后,将5至10毫克(mg)的所得样品转移到密闭式铝盘(密闭式铝盘购自ta仪器,器件编码为:ta900793-901/900794-901)。密封该盘且放置于自动样品托盘中。dsc分析的方法(本领域的技术人员已知的方法)的简要说明如下:阶段条件1在0℃下平衡2匀变5.00℃/min到250℃3标记第1循环的结束4在250℃下等温10min5在30.00℃下平衡6标记第2循环的结束7匀变以10.00℃/min匀变到250℃8方法结束动态机械热分析通过动态机械热分析(dmta),使用ta仪器变流仪(型号:ares)测定玻璃化转变温度(tg)。将矩形样品(约6.35cm×1.27cm×0.32cm)放置于固态固定装置中,并且使其经受振荡扭转负载。以3℃/min和1赫兹(hz)频率将样品从约25℃热匀变到约300℃。机械特性(拉伸和断裂韧度)使用astmd638(i型)方法进行拉伸测试。根据astmd5045通过螺杆-驱动材料测试机(英斯特朗(instron)型号5567)测量材料的断裂韧度(k1c)。使用紧凑-拉伸几何结构。实例1-cu(bf4)2和cr(c8h15o2)3加速剂的合成使用以下通用程序;以及使用在实例2和实例3中描述的用于调配物的量和化合物合成cu(bf4)2和cr(c8h15o2)3的加速剂。四氟硼酸铜(ii)(cu(bf4)2)水合物获自西格玛-奥德里奇化学公司。在70℃至80℃下在剧烈搅动下,将cu(bf4)2与cr(c8h15o2)3混合2hr至3hr。获得混合比为1:1、1:3、1:5、1:7、3:1、3:5和3:7的cu(bf4)2/cr(c8h15o2)3的各种混合物以作为用于环氧-胺组合物的加速剂进行评估。实例2-使用cu(bf4)2和cr(c8h15o2)3加速双酚a的二缩水甘油醚/芳香族胺反应包含环氧化合物、芳香族胺和本发明的加速剂的可固化组合物的实例在表i中示出。用于此实例中的加速剂为如实例1中所述制备的加速剂并且为cu(bf4)2和cr(c8h15o2)3的混合物。加速剂首先溶解于detda中,并且然后用作催化的芳香族胺(部分b)。双酚a的二缩水甘油醚(d.e.r.383)用作环氧树脂(部分a)。如表i中示出,环氧树脂和芳香族胺与加速剂以1:1化学计量比混合。表i-可固化组合物为测试在各种加速剂的情况下芳香族胺的反应性,通过dsc测量在加速剂cu(bf4)2和cr(c8h15o2)3的情况下d.e.r.383/detda的放热波峰,如图1和表ii中示出。图1示出通过dsc测量的在加速剂cu(bf4)2和cr(c8h15o2)3的情况下d.e.r.383/detda的放热峰值。“3%cr(c8h15o2)3和3%cu(bf4)2”是指在detda硬化剂中3%cr(c8h15o2)3和3wt%cu(bf4)2。在3%cu(bf4)2的情况下d.e.r.383/detda的放热峰值温度为164℃。在3%cu(bf4)2的情况下在将1%、3%、5%到7%的cr(c8h15o2)3添加到detda中之后,放热峰值逐渐从164℃偏移到112℃。放热峰值非常尖锐,表明快速固化反应。在cr(c8h15o2)3和cu(bf4)2的混合物作为加速剂的情况下,经固化d.e.r.383/detda体系的tg保持较高,在180℃到200℃的范围内。作为比较,当仅cr(c8h15o2)3用作加速剂而不添加cu(bf4)2时,放热峰值温度高达194℃并且d.e.r.383/detda在250℃下无法完全固化,意味着cr(c8h15o2)3单独无法催化反应。优异的催化活性是由于两种组分cr(c8h15o2)3和cu(bf4)2的协同效应。实例3-加速剂组分cu(bf4)2和cr(c8h15o2)3的浓度对起始温度和放热峰值温度的影响使用在芳香族胺硬化剂中的加速剂组合物进行实验设计(doe)。甚至在非常低量的加速剂(1%cr(c8h15o2)3和1%cu(bf4)2的情况下,相对于无固化加速剂的对应组合物,放热峰值温度可降低53℃(187℃到134℃,降低28%)并且起始温度可降低41℃(144℃到103℃,降低28%)。在加速剂的情况下树脂的焓和tg略微高于无加速剂的树脂。表ii-通过dsc测量的热特性对cu(bf4)2和cr(c8h15o2)3浓度*1%cu(bf4)2-1%cr(c8h15o2)3是指在detda中1wt%cu(bf4)2和1wt%cr(c8h15o2)3。实例4-调配物在各种cu(bf4)2和cr(c8h15o2)3加速剂浓度下的胶凝时间为进一步研究在加速剂的情况下树脂的时延和反应性,在25℃和100℃下测量d.e.r.383/detda在各种加速剂的情况下的胶凝时间,如表iii中示出。在25℃下在加速剂的情况下d.e.r.383/detda的胶凝时间超过30hr,这表明此树脂体系仍具有良好适用期和时延。对于在1%cu(bf4)2的情况下的d.e.r.383/detda,在100℃下的胶凝时间为96min。如果cu(bf4)2和cr(c8h15o2)3的混合物用作加速剂,那么在1%cu(bf4)2和1%cr(c8h15o2)3作为加速剂的情况下,在100℃下的胶凝时间下降到18min,并且在1%cu(bf4)2和2%cr(c8h15o2)3的情况下,下降到仅6min。在高温(150℃)下,在1%cu(bf4)2和2%cr(c8h15o2)3的情况下d.e.r.383/detda的胶凝时间仅为59s。此实例的结果进一步证实本发明的新颖加速剂可显著地改善d.e.r.383/detda的反应性。表iii-d.e.r.383/detda在各种加速剂的情况下的胶凝时间实例5-使用cu(bf4)2和cr(c8h15o2)3的环氧化合物胺化学计量对tg的影响环氧化合物的均聚通常导致tg和机械特性的下降。为确定在dsc中示出的加速的反应是否是由于环氧化合物的均聚的加速,进行以下分析:使用1%cu(bf4)2和1%cr(c8h15o2)3作为加速剂,在d.e.r.383/detda的各种化学计量比下测量经固化聚合物的tg.如果加速剂仅催化环氧化合物的均聚,那么经固化聚合物的tg将不受d.e.r.383/detda的化学计量比影响。图2示出通过1%cu(bf4)2和1%cr(c8h15o2)3催化的在d.e.r.383/detda的各种化学计量比情况下经固化聚合物的tg。通过dsc分析tg。如图2中示出,具有约1:1化学计量比的d.e.r.383/detda提供最高的tg,这支持下述结果:本发明的新颖加速剂改善环氧化合物和硬化剂之间的反应性而非加速环氧化合物的均聚。实例6-使用der383、detda和cu(bf4)2和cr(c8h15o2)3加速剂制备和机械表征透明铸件通过制备透明铸造薄片测试在新颖加速剂的情况下经固化d.e.r.383/detda体系的机械特性。固化条件包括在100℃下使树脂固化1hr以及在180℃下固化1hr。产物,具有3%cu(bf4)2)的detda用作基准(固化条件:100℃持续1hr和180℃持续2hr)。在新颖加速剂的情况下d.e.r.383/detda的拉伸模量、断裂韧度(k1c)和tg(tanδ)的特性比得上含有d.e.r.383/detda与3%cu(bf4)2作为加速剂的体系的特性,其中拉伸略微下降。总的来说,本发明的加速剂的确引起具有较高tg和高模量的经固化环氧化合物的形成。图3示出在加速剂1%cu(bf4)2和1%cr(c8h15o2)3的情况下d.e.r.383/detda的dmta。表iv-在cu(bf4)2和cr(c8h15o2)3加速剂的情况下d.e.r.383/detda的机械特性实例7-使用cu(bf4)2和cr(c8h15o2)3加速酚醛清漆树脂/detda反应在此实例7中,如实例1中所述制备的本发明的加速剂--cu(bf4)2和cr(c8h15o2)3)--用于加速芳香族胺,detda硬化剂和环氧酚醛树脂,d.e.n.431之间的反应。d.e.n.431为具有172至179的eew且可商购自陶氏化学的环氧酚醛树脂。图4示出如通过dsc测量的在加速剂1%cu(bf4)2和1%cr(c8h15o2)3的情况下d.e.r.431/detda的放热峰值。如图4中示出,当用detda硬化剂固化d.e.n.431且使用含有1%cr(c8h15o2)3和1%cu(bf4)2的加速剂时,相较于仅有1%cu(bf4)2作为加速剂的对应树脂组合物,加速剂的使用可使树脂组合物的放热峰值温度降低45℃(从176℃到131℃,降低26%)。实例8-zn(bf4)2和cr(c8h15o23)加速剂的合成使用以下通用程序;以及使用用于在下文实例9中描述的调配物的量和化合物合成zn(bf4)2和cr(c8h15o23)的加速剂。四氟硼酸锌(ii)(zn(bf4)2)水合物和二乙二醇获自西格玛-奥德里奇公司。在25℃下在剧烈搅动下,将zn(bf4)2与cr(c8h15o2)3以及二乙二醇混合2min。在加速剂体系中zn(bf4)2、cr(c8h15o2)3和二乙二醇的重量百分比分别为40%、40%和20%。制备的zn(bf4)2和cr(c8h15o2)3/二乙二醇的混合物用作用于环氧-芳香族胺反应的加速剂。在此实例8中,zn(bf4)2和cr(c8h15o2)3为加速剂必需的组分;以及二乙二醇为载体溶剂。实例9-使用zn(bf4)2和cr(c8h15o2)3加速剂加速双酚a的二缩水甘油醚/detda反应在此实例9中,四氟硼酸锌(ii)(zn(bf4)2)与cr(c8h15o2)3一起用作加速剂。如实例8中所述制备加速剂。图4示出通过dsc测量的在加速剂1%zn(bf4)2和1%cr(c8h15o2)3的情况下d.e.r.383/detda的放热峰值。如图5中示出,相较于不含催化剂或单独1%zn(bf4)2的对应树脂调配物,含有1%zn(bf4)2和1%cr(c8h15o2)3的加速剂可显著地加速环氧-芳香族胺反应。实例10-使用zn(bf4)2和cr(c8h15o2)3加速剂加速双酚a的二缩水甘油醚z反应除了二乙基甲苯二胺(detda)之外,还测试了其它芳香族胺,其中一个实例示出为z。z为芳香族胺(1,3-苯二胺和4,4'-二氨基二苯基甲烷)的共混物且可商购自空气产品。图6示出通过dsc测量的在加速剂1%zn(bf4)2和1%cr(c8h15o2)3的情况下d.e.r.383/z的放热峰值。加速剂(1%zn(bf4)2和1%cr(c8h15o2)3)可使树脂组合物的放热峰值温度降低44℃(从142℃到98℃,降低31%),以及使树脂组合物的起始温度降低46℃(从101℃到55℃,降低45%)。实例11-使用zn(bf4)2和cr(c8h15o2)3加速剂加速双酚a的二缩水甘油醚d230(聚醚胺)反应在此实例11中,使用在实例8中示出的程序;以及使用如下描述的用于调配物的量和化合物合成zn(bf4)2和cr(c8h15o2)3的加速剂。包含环氧化合物、聚醚胺和本发明的加速剂的可固化组合物的实例在表vi中示出。在此实例中使用的加速剂为如中实例8所述制备的加速剂并且为zn(bf4)2和cr(c8h15o2)3的混合物。用于此实例中的聚醚胺为d230,其可商购自亨茨曼公司。加速剂首先溶解于聚醚胺中,并且用作催化的聚醚胺(部分b)。双酚a的二缩水甘油醚(d.e.r.383)用作环氧树脂(部分a)。如表vi中示出,环氧树脂和聚醚胺与加速剂以1:1化学计量比混合。测量在有和没有加速剂(zn(bf4)2和cr(c8h15o2)3)的情况下d.e.r.383d230在100℃下的胶凝时间和放热峰值以测试反应活性。如表vii中示出,加速剂(zn(bf4)2和cr(c8h15o2)3)可将在100℃下的胶凝时间从7min30sec缩短到3min50sec。同时,加速剂可使放热峰值温度降低15℃(从129℃到114℃,降低11.6%)并且使起始温度降低25℃(从89℃到65℃,降低28%)。表vi-可固化组合物表vii-通过dsc测量的在加速剂2%zn(bf4)2和2%cr(c8h15o2)3的情况下der383d230的胶凝时间和热特性实例12-zn(bf4)2和cr(acac)3加速剂的合成在此实例12中,使用以下通用程序;以及使用用于如下描述的调配物的量和化合物合成zn(bf4)2和cr(acac)3的加速剂。除了cr(c8h15o2)3之外,还测试了其它羧酸铬(iii),其中乙酰基丙酮酸铬作为一个实例。乙酰基丙酮酸铬(iii)(cr(acac)3)可商购自西格玛-奥德里奇化学公司。四氟硼酸锌(ii)(zn(bf4)2)水合物、乙酰基丙酮酸铬(iii)(cr(acac)3)和二乙二醇获自西格玛-奥德里奇公司。在25℃下在剧烈搅动下,将zn(bf4)2与cr(acac)3和二乙二醇混合1hr。在加速剂体系中zn(bf4)2、cr(acac)3和二乙二醇的重量百分比分别为33.3%、33.3%和33.3%。制备的zn(bf4)2、cr(acac)3和二乙二醇的混合物用作用于环氧-芳香族胺反应的加速剂。在此实例中,zn(bf4)2和cr(acac)3为加速剂必需的组分并且二乙二醇为载体溶剂。实例13-使用zn(bf4)2和cr(acac)3加速d.e.r.383/detda反应1%zn(bf4)2和1%cr(acac)3组合用作用于d.e.r.383/detda反应的加速剂。表viii示出如通过dsc测量的在没有加速剂(对照组)和有加速剂1%zn(bf4)2和1%cr(acac)3的情况下d.e.r.383/detda的热特性。加速剂(1%zn(bf4)2和1%cr(acac)3)可使树脂组合物的放热峰值温度降低39℃(从187℃到148℃,降低21%),以及使降低树脂组合物的起始温度降低67℃(从144℃到77℃,降低46.5%)。表viii-通过dsc测量的在加速剂1%zn(bf4)2和1%cr(acac)3的情况下d.e.r.383/detda的热特性如以上实例中示出,用于环氧-胺树脂的含有羧酸铬(iii)(例如cr(c8h15o2)3)和盐(其中盐的阳离子为金属阳离子或带有非亲核性阴离子的鎓阳离子(例如cu(bf4)2或zn(bf4)2))的本发明的新颖加速剂可大大加速环氧化合物和芳香族胺之间的反应,同时维持经固化材料的优异机械和化学特性。本发明的加速剂可显著缩短固化特性曲线并且当胺用作硬化剂时降低处理成本,以及使胺硬化剂的应用领域变宽。实例14-在der383和detda反应中不使用没有加速剂,仅cu(bf4)2或cu(bf4)2与cr(c8h15o2)3、zn(c8h15o2)2、mo(c8h15o2)3组合比较峰值放热温度在现有技术中已知,实验条件例如dsc扫描速率可影响所得产物的tg和放热起始温度。在本发明的研发期间,使用5℃/min扫描速率。然而,由于在现有技术中常见以10℃/min扫描速率进行dsc实验,出于比较目的,所以此实例描述并比较以10℃/min扫描速率测定的放热峰值温度。使用如中表i所述以1:1化学计量混合比的der383和detda。在detda中以在表ix中列出的浓度混合加速剂。相较于未催化的体系,单独cu(bf4)2仅造成峰值放热温度偏移17%。然而,相较于未催化的体系,cu(bf4)2和各种辛酸盐的组合的使用造成峰值放热显著的偏移,即对于cr(c8h15o2)3偏移42%,对于zn(c8h15o2)3偏移25%,以及对于mo(c8h15o2)3偏移25%。但是,如果放热是相较于单独用cu(bf4)2催化的反应,那么峰值放热的偏移也是值得注意的。在这种情况下,峰值放热的偏移对于cr(c8h15o2)3为30%,对于zn(c8h15o2)3为10%,以及对于mo(c8h15o2)3为9%。这些偏移说明使用cu(bf4)2和辛酸盐的组合产生比单独cu(bf4)2快的加速剂。此实例的结果描述于表ix中。表ix-通过dsc10℃/min扫描速率测定的使用各种加速剂的d.e.r.383和detda的固化反应的峰值放热温度的比较实例15-在150℃固化温度下通过cu(bf4)2或cu(bf4)2和cr(c8h15o2)3催化的der383-detda体系的tg的比较der383和detda以如表i中所述的1:1当量比混合。此混合物以两种不同方式催化,一种用0.6%cu(bf4)2和另一种用0.6%cu(bf4)2和2%cr(c8h15o2)3的组合,其中百分比是相对于总组合物而言。将der383-detda-加速剂混合物的10g部分放置于铝盘中并且在150℃烘箱中固化5min、10min和60min。通过dsc(10℃/min扫描速率)测量经固化热固性材料的tg并且在表x中列出。如在表x中的数据示出,cu(bf4)2和cr(c8h15o2)3的组合使用显著加速固化反应,并且甚至在60min固化时间之后提供相较于单独使用cu(bf4)2高57℃的tg。为达到相同tg,在单独cu(bf4)2的情况下将需要长得多的固化时间或高得多的温度。表x-在150℃下固化不同时间的der383-detda热固性材料的tg固化时间(分钟)51060玻璃化转变温度tg(℃)tg(℃)tg(℃)0.6%cu(bf4)251941090.6%cu(bf4)22%cr(c8h15o2)3144148166当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1