微粒的制作方法
本发明涉及微粒,具体而言涉及自组装微粒,制备所述微粒的方法,制备通过所述颗粒的碰撞形成的大孔材料,以及所获得的颗粒和多孔结构的用途。微粒和多孔材料可用于广泛的物理和化学过程,特别是需要与底物相互作用的那些,例如固相合成、固相萃取、固相反应剂、物质(例如蛋白质和核酸)的固定化、细胞培养、伤口护理包括内部和外部慢性伤口和急性创伤、烧伤的治疗、医学诊断、再生医学、视力矫正、化学品如药品和农用化学品的控制释放、催化和色谱。
用于固相合成过程的固体支持材料是已知的。大量的物理和化学过程采用固体支持材料,包括例如有机分子(特别是肽和寡核苷酸)的合成、物质的固定化、催化剂的支持、离子交换、从材料中萃取物质、诊断和色谱。
通常,有机分子的多阶段合成涉及在进行到后续阶段前的许多分离步骤,以分离在每个阶段产生的中间体。这些过程常常是耗时且昂贵的,并且对于产率而言是低效的。中间体常常需要经过纯化以除去过量的反应剂和反应副产物,并需要例如沉淀、过滤、双相溶剂萃取等程序;可以采用固相萃取、结晶和色谱法。
固相合成比溶液相合成提供了一些优点。例如,可以通过将靶分子可逆地附着到固体支持物上,以在某种程度上避免在溶液相合成中用到的分离程序。可以通过过滤和洗涤固体支持物来除去过量的反应剂和一些副产物。可以在一些过程中以基本上定量的产率回收靶分子,这在溶液相合成中通常特别困难。此外,在固体支持物上进行操作所需的时间通常远小于在溶液相合成中进行等效阶段所需的时间。
在一系列过程中对物质的固定化也是已知的。例如,聚合物支持物通常用于固定化用于传统有机化学(包括化学和生物催化)的催化剂。可以使用固定化的酶来进行有机化学反应或用于手性拆分,例如使用固定化的青霉素酰胺酶来分解仲醇(e.baldaro等人,tet.asym.4,1031,1993)以及还可以使用固定化的青霉素g酰胺酶在阿莫西林制造中进行苄青霉素的水解(carleysmith,s.w.和lilly,m.d..biotechnol.bioeng.,21,1057-73,1979)。
固体支持物也可用于固定化生物大分子以用于医疗和诊断应用。这包括蛋白质、单克隆抗体和多克隆抗体的固定化。细胞培养通常在具有特定表面特征和形态的固体支持物上进行。固定化的酶可用作传感器以生成信号。一个实例是通过葡萄糖氧化酶/过氧化物酶偶联的酶系统来检测葡萄糖,其中葡萄糖的存在产生过氧化氢,其又是过氧化物酶的底物,过氧化物酶用于氧化多种底物以提供有色的、荧光的或发光的信号。
其荧光对特定阳离子或阴离子敏感的多种染料(fluor)可用于指示具体离子的浓度,包括用于ph测量的氢离子。
聚合物颗粒和多孔材料常常用于色谱中,其中固体支持物称为固定相。在某些色谱模式下,固定相的成本可能产生一些限制。在其它模式中,固定相的物理性质可降低该技术的有效性。例如,常常用于亲和力、离子交换和凝胶渗透色谱的软聚合物不能以高流速使用,因为颗粒的可变形性质。用于许多其它色谱模式的刚性大孔聚合物经常是机械易碎的,并因此受到寿命短的困扰。
固相支持物或固定相在色谱分离中的应用非常广泛,例如在制药和生物工业中使用的复杂的高技术分离以及在采矿工业中使用的更大规模的过程。一些制药工业中最有价值的药品是通过制备色谱法纯化的,而改进的色谱分离能取得技术上的益处和经济上的优势。在采矿和贵金属回收工业中,可以使用固定化的冠醚来精炼世界上大部分的钯,其是广泛范围的工业应用和过程(包括催化转化器和高价值产品的制造)中的关键组分(traczyk,f.p.;bruening,r.l.;izatt,n.e.“theapplicationofmolecularrecognitiontechnology(mrt)forremovalandrecoveryofmetalionsfromaqueoussolutions”;infortschritteinderhydrometallurgie;1998,
聚合物颗粒和大孔材料在固相萃取中和在固相反应剂的制备中的用途在化学、制药和生物技术工业也是已知的。
已知的固相支持物通常包含特定尺寸和物理性质的聚合物颗粒,以适应应用。为了易于使用,这些聚合物颗粒常常是球形的并且具有确定的颗粒尺寸分布。颗粒的球形性质改善了聚合物的流动和过滤特性。虽然固体支持物的使用具有操作优势,但固相方法存在缺点。例如,通常用于肽和寡核苷酸的固相合成的市售支持物可能昂贵,例如是由于复杂的制备过程。通常使用的是微孔聚合物颗粒和大孔聚合物。微孔聚合物具有相对低水平的交联剂,其允许聚合物颗粒在合适的溶剂中溶解并随后膨胀。大孔聚合物在聚合物基质中具有高水平的交联剂并含有大孔。这些聚合物颗粒通常是刚性的并且具有良好的流动特性,适用于填充柱。
伤口处理和伤口护理是一个重大的临床挑战。许多已知的伤口护理处理使用动物衍生的胶原,例如牛、马、猪和人衍生的胶原。这些材料的使用可能会因伦理、道德和宗教原因而出现一系列挑战,这些挑战阻碍了其的广泛使用。动物衍生的胶原也可能呈现技术和商业问题,因为这些产品的提取和加工可能是复杂、缓慢和昂贵的生产。在需要动物衍生产品的某种特定来源时,例如某些商业产品依赖于马腱的使用,供应可能难以满足。动物衍生产品的生产可以产生生物废物,并需要特别的处理以降低生物污染的风险。
需要适用于伤口护理应用的材料,其不含有源自动物的组分,但是其是生物相容性的、可生物降解的、非细胞毒性的并理想地是抗菌的。
在许多的这些使用领域中使用的已知聚合物颗粒具有某些缺陷。聚合物颗粒通常可以通过分散或乳化聚合过程来制备,其中在聚合开始之前将单体溶液分散在不混溶溶剂(连续相)中。然后通常将形成的聚合物颗粒过滤、洗涤和分级以分离所需的颗粒尺寸分布。然而,该过程可能复杂且昂贵,并且受到需要使用有机溶剂的限制。本文所用的术语“聚合物”包括无机聚合物,例如二氧化硅,以及有机聚合物,例如聚酰胺。
这些过程在某些方面是不利的,包括单体损失在连续相中、产生一定范围的颗粒尺寸,以及在聚合过程中产生不期望的细颗粒导致颗粒尺寸分级困难,例如通过筛分或空气分级。
除了生产中不期望的成本和在制备过程中的浪费之外,已知的聚合物颗粒的物理性质可能会产生某些缺点。微孔聚合物颗粒通常是软的,并且通常不适合用于在填充柱床中以高流速进行色谱应用。此外,软颗粒可能受到不期望的压缩并引起结垢,例如在过滤期间常常导致挤压进入在柱底部使用的烧结物或网状物中。刚性大孔和大网状颗粒更适合于在填充柱床中的高流速。然而,由于刚性,颗粒在物理应力下可能变脆或破碎。
在将聚合物颗粒从底部向上填充到柱中时,聚合物颗粒受到不期望的大应力,此时这些问题则会加剧。
晶球通常包含多层表面活性剂作为水包油组合物,并且可能需要使用溶剂和复杂的温度控制来形成晶球。颗粒尺寸可以在很宽的范围内延伸。
我们现在已经发现,可以通过提供颗粒支持物来改善与已知的聚合物颗粒、大孔材料和已知晶球相关的这些和其它问题,所述颗粒支持物包括自组装微粒或大孔材料,其中自组装微粒包含具有两个或更多个羧酸基团的脂肪酸和提供窄的颗粒尺寸分布的碱,大孔材料将自组装微粒相接触而形成。
在第一方面,本发明提供包含自组装微粒的颗粒支持物。适当地,所述微粒包含具有两个或更多个酸基的酸和溶于亲水性溶剂的有机碱。
优选地,所述酸包括双酸,优选地双脂肪酸,并且适当地包含两个或更多个羧基基团,尽管也可以使用其它酸基。适当地,所述双酸不溶于或微溶于亲水溶剂。适当地,通过使所述酸,优选地双脂肪酸与溶于亲水溶剂的有机碱接触,可以将酸溶解。
适当地,溶剂是亲水性的,优选地水性溶液,例如水相中的油包水乳液,并且尤其是水。有利地,水基溶剂,优选地水,允许微粒用于其中对环境的考虑很重要的应用中。例如,可以将微粒配制成水基产品,其可适用于个人使用或消费、医疗用途,例如作为杀生物剂。已知的杀生物剂可以含有环境不期望的溶剂或组分,例如需要小心使用和处置以及后续清洁的异丙醇和硅氧烷,例如当用于清洁或抗微生物应用时。本发明提供的水基组合物减少或避免了与含有有机溶剂或硅酮的产品相关的缺点。
在优选的实施方案中,所述双脂肪酸包括双羧酸脂肪酸,其中末端羧酸通过比末端羧酸亲水性更低的(优选是疏水性的)区域连接。亲水性更低的区域可以包含具有取代物的骨架,和/或骨架可以包含杂原子,例如聚ε-赖氨酸。优选地,连接羧酸的区域是疏水性的并且优选烃基。在特别优选的实施方案中,疏水基团是脂族烃基。优选地,双酸包括通式为hooc-(ch2)n-cooh的化合物,其中n足够大,使得双酸微溶或不溶于水。n优选地为至少5、更优选地至少6、特别是至少7。适宜地,n不大于40、优选地不大于36、更优选地不大于25、并且特别是不大于20。n优选地为7至18。
在优选的实施方案中,有机酸包括c7至c18双羧酸脂肪酸。在另一个优选的实施方案中,有机酸包括c7至c13双羧酸脂肪酸,以及另外的酸,其选自edta、硝基三乙酸和一元羧酸,优选地c6至c18羧酸,例如己酸、棕榈酸和辛酸。
通过选择多于一种酸,例如其中酸具有不同的n值,可以调整微粒的尺寸。连接酸基的更长疏水部分适当地提供更大的微粒。例如,当n为8时,为癸二酸,可以获得尺寸为2.6微米的颗粒,并且其中n为11时,为巴西基酸,可以获得大小为3.0微米的颗粒。
双羧基脂肪酸也可以是不饱和的(例如愈伤酸),或取代的,或者不饱和且取代的。适当地,取代不会导致双酸溶于水性溶液。当双脂族酸在溶剂可溶性有机碱的帮助下接触时,自发形成微粒。
双脂族酸可以包括:通式(ho)2op-(ch2)n-po(oh)2的双膦酸或不饱和双膦酸;通式为hooc-(ch2)n-po(oh)2的单羧酸单膦酸或这种双酸的不饱和形式;通式(ho)o2s-(ch2)n-so2(oh)的双磺酸或这种双酸的不饱和形式;通式为hooc-(ch2)n-so2(oh)的单羧酸单磺酸或这种双酸的不饱和形式;通式(ho)2b-(ch2)n-b(oh)2的双硼酸或不饱和双硼酸或取代的双硼酸;通式为hooc-(ch2)n-b(oh)2的单羧酸单硼酸或这种双酸的不饱和形式;或者所述双酸的取代形式。在这些酸中,n足够大,使得双酸微溶或不溶于水。n优选地为至少5、更优选地至少6、并且特别地至少7。合适地,n不大于40、优选地不大于36、更优选地不大于25、并且特别是不大于20。n优选地为7至18。
合适地,有机碱与双酸部分组合,使得两种组分的组合包含两个分开的亲水的或离子的头部区域,它们通过疏水区连接。不希望受理论束缚,据信相邻双酸的疏水区域和亲水区域与有机碱对齐时形成胶束并导致本发明的微粒的自组装。优选地,微粒包含多层结构,其中包含双酸与有机碱的其它分子与另一双酸/有机碱的亲水头对齐,以形成多层结构。
有机碱可以选自与双酸一起形成自组装微粒的一系列碱。优选地,有机碱包括胺,适当地,是具有碱性特征或其它含氮碱的脂族胺或芳香族胺。合适的有机碱的实例包括烷基化胺和多胺,包括具有一个或两个c1-4n-烷基基团的胺,例如甲基化胺。优选的胺的实例包括n-甲基吗啉、4-甲基吗啉(nmm)、n,n-二甲基氨基乙醇(dmae)、4-二甲基氨基吡啶(dmap)、咪唑或1-甲基咪唑、聚(二烯丙基二甲基氯化铵)(pdac)、二癸基二甲基氯化铵(ddac)和十二烷基二亚丙基三胺(ddpt)。
在优选的实施方式中,酸合适地是巴西基酸、癸二酸和壬二酸中的一种或多种,与选自甲基吗啉(nmm)、n,n-二甲基氨基乙醇(dmae)、4-二甲基氨基吡啶(dmap)、咪唑、1-甲基咪唑、聚(二烯丙基二甲基氯化铵)(pdac)、二癸基二甲基氯化铵(ddac)和十二烷基二亚丙基三胺(ddpt)的碱组合。
优选的实例包括微粒,其包含巴西基酸和pdac,巴西基酸和ddac,巴西基酸和ddpt,癸二酸和nmm,聚ε-赖氨酸与癸二酸,巴西基酸和壬二酸中一种或多种的组合。
我们已经发现根据本发明的包含具有抗微生物性质的胺的微粒特别适合用作抗微生物组合物和杀生物剂。当以根据本发明的自组装微粒的形式时,碱的抗微生物活性水平可以比常规剂型更高。
根据另一方面,本发明提供了抗微生物组合物,其包含自组装微粒,所述自组装微粒包含双酸和抗微生物碱。本发明还提供了自组装微粒的用途,所述自组装微粒包含双酸和抗微生物碱,其比所述抗微生物碱的非自组装微粒形式具有更高的抗微生物活性水平。
适当地,提供在自组装微粒中的抗微生物碱增加了抗微生物活性并且提供至少2log的细菌载量减少、优选地至少4log的细菌载量减少,理想地至少5log的细菌载量减少。
以使得酸中的酸基与碱中的碱基的摩尔比为形成自组装微粒的大约化学计量的相对量,将酸和碱适当地组合。只要形成自组装颗粒,酸基与碱基的摩尔量可以小于或大于化学计量。当酸基与碱基的比例太低或太高时,过量组分破坏酸和碱的结构而不会形成组装的颗粒。允许形成自组装颗粒的酸基与碱基的比例将根据具体的酸和具体的碱而变化。
本领域技术人员将能够通过在显微镜下以能目视观察颗粒的水平的放大率(例如以40倍放大率)观察来确定自组装颗粒是否形成。能够修饰酸和碱的相对量,以确定形成微粒的组分的最小和最大比例。具有更长链的酸可以提供比包含具有更短链的酸的微粒(具有相同的碱和相同的摩尔比)更稳定的微粒。更大的稳定性可以允许采用更低水平的酸,并且酸基与碱基的更低比例可能仍然允许微粒形成。
适当地,酸和碱中酸基与碱基的比例为0.6至1.4:1、优选地0.7至1.3:1、更优选地0.8至1.2:1、并且理想地为0.9至1.1:1。癸二酸和巴西基酸是优选的酸的实例。适当地,包含癸二酸与碱的微粒具有癸二酸与碱的比例为0.85至1.15:1。包含巴西基酸与碱的微粒具有巴西基酸与碱的比例为0.8至1.2:1。在一个优选的实施方案中,酸和碱以提供1:1的酸基与碱基的摩尔比的水平存在。
在第二方面,本发明提供了通过在诸如形成大孔材料的条件下使自组装微粒接触而形成的大孔材料。适当地,大孔材料通过微粒交联而适当地形成。
有机碱可以是反应性的,以便能够使自组装微粒交联以形成大孔材料。有机碱并不需要是反应性的,在这种情况下,它可以适当地被另一种反应性物质置换,以允许随后交联而形成大孔材料。溶剂可溶的有机碱可以通过加入反应性物质而被置换,反应性物质包括但不限于含胺的有机组分。适当地,胺允许通过酰胺键形成来交联微粒。在优选的实施方案中,含胺有机组分是聚合胺,包括但不限于肽、蛋白质、聚烯丙胺、聚乙烯亚胺和其它多胺。
合适的胺和多胺的实例包括乙二胺、聚-ε-赖氨酸、聚烯丙胺、聚乙烯亚胺、氨基丙基三烷氧基硅烷、3-(2-氨基乙基氨基)丙基三甲氧基硅烷、n-(3-(三甲氧基甲硅烷基)-丙基)二乙基三胺。
在形成微粒或大孔材料时,上述双酸可以以任何比例混合。此外,也可以混合反应性胺。
适当地,微粒或大孔材料包含根据预期用途定制的功能组分。例如,加入乙二胺四乙酸赋予金属螯合特性。
在另一个实施方案中,聚乙烯亚胺可用于结合或者作为合成或操作例如对核酸包括dna、rna的测序中的支持结构。
可以使用烷氧基硅烷,并且可以在微粒的层状层中形成二氧化硅壳。
在另一个实施方案中,可以将特异性酶的活性位点并入到颗粒内的肽中以允许活性剂的控制释放。例如,可以将基于伤口的金属蛋白酶的切割位点并入基于伤口护理的材料中以允许抗细菌剂的控制释放。
在另一个应用中,本发明的微粒可用于形成用于骨细胞生长的宏观结构。适当地,微粒与羟基磷灰石组合,优选地以结晶形式,以吸附骨细胞并促进骨细胞培养。
现在的许多市售伤口敷料包含动物衍生的胶原,其已被证明可以改善伤口愈合。本发明的微粒和大孔材料可用于替代伤口敷料中的动物衍生胶原。已经显示本发明的大孔材料在组织修复中模拟胶原的生物学和物理学性质。然而,这些材料的组分不会背负在人类医疗保健中使用基于动物的材料所考虑的越来越多的道德和宗教问题。
根据本发明的自组装微粒或大孔材料还可以包含由聚合物支持的功能材料。合适的功能材料的实例包括催化剂、用于肽合成或寡核苷酸合成的引发剂物质、药物活性物质、农业化学活性物质、大分子、酶、核酸序列和蛋白质。
本发明特别可用于支持贵金属催化剂,例如钯催化剂。一个特别有利的实例是钯。
本发明在另一方面提供了用于在水性介质中产生自组装微粒或大孔材料的方法,包括在水性介质优选地水中使具有两个或更多个酸基的两种酸与有机碱接触。
适当地,通过本领域技术人员已知的过程引发聚合和交联。例如,可以使用水溶性碳二亚胺将在水中制备的自组装微粒或大孔材料与含有胺的组分进行交联。
本发明的自组装微粒或大孔材料可以用于其中使用固体支持物的任何化学或物理过程。
所述自组装微粒或大孔材料可用于涉及导电和发光聚合物的应用中。可以将含有发光聚合物的颗粒支持物布置在显示面板上。
所述自组装微粒或大孔材料特别适用于有机物质,特别是大分子的固相合成。在优选的实施方案中,自组装微粒或大孔材料可用于肽、寡核苷酸或寡糖的合成。
在固相合成中,在诸如肽合成的应用中使用固体聚合物颗粒的过程通常包括将颗粒悬浮在多孔过滤板上方的合适溶剂中并轻轻搅拌颗粒从而不机械地损坏颗粒。颗粒的制造过程常常产生细粉,引起过滤板堵塞,导致缓慢过滤或需要更换或清洁过滤器。此外,固体颗粒的搅拌可能引起断裂,导致细粉的产生,这加重过滤器堵塞的问题。在制药和相关行业中,动态药品生产管理规范(cgmp)下严格的质量法规要求在每批产品之后更换过滤板,以避免随后的批次被从过滤板上移出的物料污染。
适当地,根据本发明的自组装微粒或大孔材料大体上是单分散的。也就是,所述材料具有全体大体上相同尺寸的颗粒。单分散微粒或大孔材料有利地简化了固相合成。
本发明进一步提供了根据本发明的自组装微粒或大孔材料作为色谱过程中的固相的用途。
通常,通过在合适的溶剂中制备颗粒的浆料或固定相并将其转移到具有底部柱过滤板的柱中对色谱柱进行填充。由于色谱柱中宽颗粒尺寸分布导致的色谱床的不均匀沉降可引起固定相床的不均匀甚至出现裂纹,导致不佳且不可再现的分离。柱子常常必须被清空并重新填充数次以达到所需的性能。这可能很费力并导致停机,这在过程的大规模操作中是一个尤其显著的缺点。
在本发明的优选实施方案中,自组装微粒几乎单分散的性质允许制备浆料并将浆料转移到柱中以形成更均匀的床。备选地,通过自组装微粒的碰撞形成的大孔材料可以用于制备整料的色谱柱。
在另一个实施方案中,整料(monolith)中颗粒之间的间隙可以用不同的组分例如细胞培养物养分填充。在该实施例中,可以在自组装的大孔材料的表面上培养细胞。在该实施例中,自组装的大孔材料常常被称为用于三维细胞培养的支架。因此,本文所述的材料将应用于再生医学、3d细胞培养和伤口护理中。
本发明的自组装微粒和大孔材料也可用于固相萃取以从与支持物接触的液体中除去物质,无论是分批形式还是作为流过支持物的流体,例如离子萃取和离子交换。固相萃取通常在带过滤板的柱中或系统中进行,所述过滤板用于将固相与进行萃取的混合物分离。在本文中提及的对固相合成和色谱所观察到的问题在固相萃取中也可类似地观察到。本发明的自组装微粒和大孔材料提供了色谱和固相合成所提供的类似的优点。
本发明的自组装微粒和大孔材料可用于固定化包括抗体、寡核苷酸、酶或染料(fluor)的物质,并且可以将其定位在阵列中,其中每种支持物测定溶液的不同组分。具有共价连接在其表面上的配体的自组装微粒和大孔材料可以用作“孔”。然后可以使用确定的方法检测靶配体如抗原或者互补dna或rna序列的特异性结合。
本发明的自组装微粒和大孔材料也可用于固定化生物催化剂。生物催化剂常常用于具有过滤板的柱中或系统中,所述过滤板用于将固相与进行萃取的混合物分离。在本文中提及的对固相合成和色谱所观察到的问题在固相萃取中也可类似地观察到。
本发明的自组装微粒和大孔材料特别可用于固定化物质,包括固相反应剂、金属和其它催化剂、生物催化剂、酶、蛋白质、抗体包括多克隆和单克隆抗体、全细胞和聚合物。本发明特别有利于支持酶,例如通常用于洗涤剂和个人护理产品中的脂肪酶calb。
本发明还特别可用于亲和配体如蛋白质a的固定化。
在另一个应用中,本发明的颗粒支持物也可以用于化学催化,例如通过固定化过渡金属催化剂和配体。
在另一个应用中,本发明可以用于细胞培养。动物细胞系的大规模培养是制造病毒疫苗、生物药物和许多生物技术产品的基础。通过重组dna技术在动物细胞培养物中生产的生物学制品包括酶、合成的激素、免疫生物学制品(单克隆抗体、白细胞介素和淋巴因子)和抗癌剂。在细菌培养物中使用rdna可以生产很多更简单的蛋白质;目前必须在动物细胞中制备糖基化(碳水化合物修饰)的更复杂的蛋白质。这种复杂蛋白质的一个重要实例是激素促红细胞生成素。培养哺乳动物细胞培养物的成本高,因此公司不断寻求改进技术。
细胞可以在悬浮中或作为贴壁培养物进行培养。然而,贴壁细胞需要表面,其可以用细胞外基质组分涂覆以增加贴壁性质并提供生长和分化所需的其它信号。通常来自固体组织的细胞是贴壁的。器官型培养包括在三维环境中培养细胞,而不是二维培养皿。这种3d培养系统在生物化学和生物学上更类似于体内组织,但是出于许多因素(例如扩散问题)在技术上难以维持。
另一方面,本发明提供了使用根据本发明的自组装微粒和大孔材料以在支持物表面上培养细胞的用途。适当地,干细胞可以在本发明的自组装微粒和大孔材料上培养,以减少不受控制的分化并控制所需的分化。自组装微粒和大孔材料的处理性质以及支持物表面积的高利用率在本申请中是有利的。
本发明在诸如免疫测定的医学诊断测试中特别有用。因此,本发明进一步提供了医学诊断,用于检测包含根据本发明的自组装微粒和/或大孔材料的化合物以及功能材料的存在情况,所述功能材料例如酶,如辣根过氧化物酶,其由支持物中的聚合物支持,与要检测的化合物选择性发生反应或与之结合。
本发明的自组装微粒和/或大孔材料可用于分离过程,例如磁性分离,流式细胞术,药品递送以及广泛的领域,包括洗涤剂,农用化学品和个人护理领域。
许多医学诊断依赖于固体支持物以固定化各种诊断配体。本发明的自组装微粒和/或大孔材料可以用于医学诊断程序,其中在液相中对固相进行物理分离。
在另外的应用中,自组装微粒和/或大孔材料可以用作吸收剂。在此应用中,如果支持物包含与自组装微粒和/或大孔材料结合的惰性吸收材料,则是特别有利的。自组装微粒和/或大孔材料可以用于吸收居家溢出物,例如茶、咖啡和葡萄酒,或者可以用于更大规模的应用中,例如从溢出物吸收油。吸收剂支持物可以用于吸收溢出物,然后物理地去除,或者在水体中有油溢出物的情况下,有效地捕获油并将油保留在要保留的物料中进行收集和处置。
本发明的自组装微粒和/或大孔材料可以用作载体以携带在一段时间内要释放的化合物,例如药物或农用化学品或组合物。该用途提供了根据在支持物上化合物的加载量来调整化合物的剂量方案的方法。在药物的情况下,这对于帮助活性物质的正确剂量是有利的,例如连续缓慢释放而不是要求患者定期服用大剂量,例如在化疗中。在非处方药的情况下,微粒可用于递送鼻解充血剂、防腐剂和抗炎辅助剂。在某些情况下,已经使用微粒来递送天然油如薄荷油和薰衣草油作为抗打鼾助剂。
除了吸入之外,自组装微粒可以以合适的形式用于静脉内药品递送或用于递送疫苗。
本发明的微粒或大孔材料可以配制成用于广泛用途的组合物,包括用于个人护理产品的组合物例如局部组合物和口腔组合物,家庭护理产品和例如伤口治疗产品的医疗产品。个人护理产品的实例包括洗手液、手磨砂膏、霜剂、除臭剂、洗发水和护发素。家居护理产品的实例包括抗微生物产品、表面喷雾清洁剂、洗涤剂等。局部组合物可以包含适于局部递送的功能材料。
通过以下非限制性实施例说明本发明。
实施例1-自组装微粒的制备
将巴西基酸(1.54g,6.31mmol)和4-二甲基氨基吡啶(dmap,1.54g,12.62mmol)溶于水(10cm3)中,并将样品置于显微镜上。观察到直径~3μm的几乎都为单分散的球形实体(图1)。
实施例2-自组装微粒的制备
将巴西基酸(1.54g,6.31mmol)和二甲基氨基乙醇(dmae,1.12g,12.62mmol)溶于水(10cm3)中,并将样品置于显微镜上。观察到直径~3μm的几乎都为单分散的球形实体。
实施例3-自组装微粒的制备
将巴西基酸(1.54g,6.31mmol)和4-甲基吗啉(nmm,1.275g,12.62mmol)溶于水(10cm3)中,并将样品置于显微镜上。观察到直径~3μm的几乎都为单分散的球形实体。
实施例4-自组装微粒的制备
使用一系列酸和一系列水溶性有机碱进行上述二羧酸溶解实验。下面列出了一些经测试的组合。这些组合具有酸基与碱基的摩尔比为0.9至1.1:1。所有这些组合都形成了球形实体,如实施例1所述。
庚二酸加nmm
辛二酸加nmm
壬二酸加nmm
癸二酸加nmm
癸二酸加dmap
癸二酸加dmae
癸二酸加咪唑
十二烷二酸加nmm
十二烷二酸加dmap
十二烷二酸加dmae
c36二聚酸加nmm
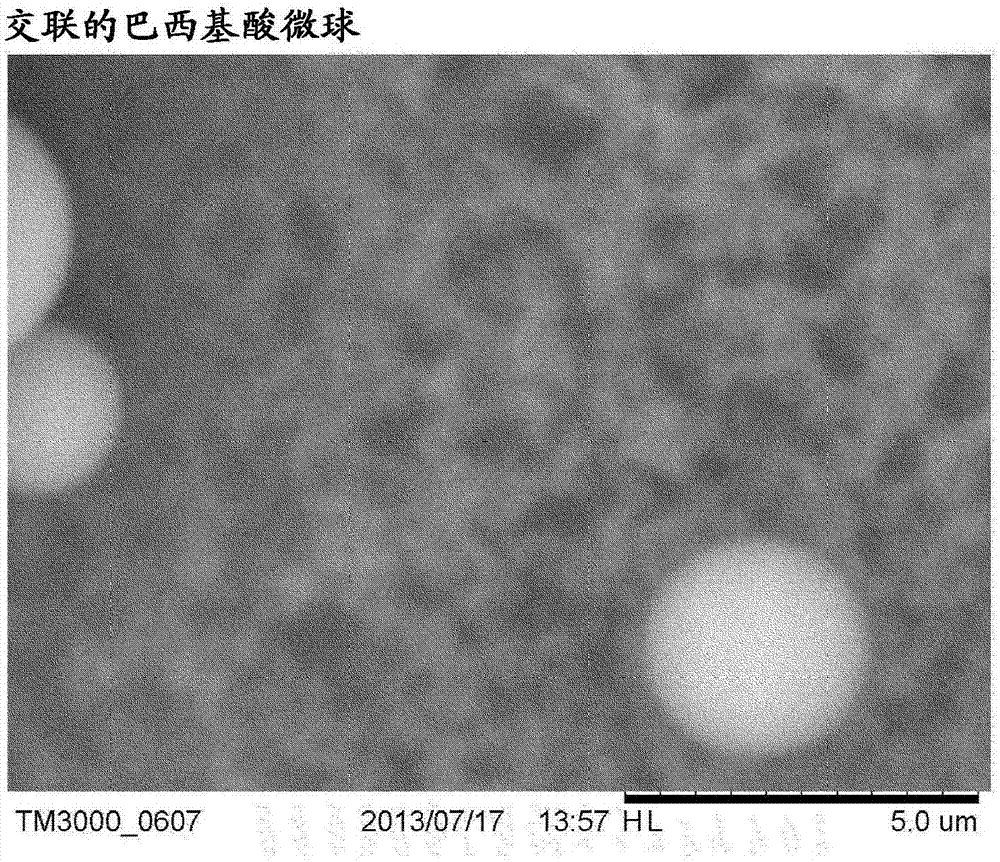
实施例5-交联的自组装微粒的制备
将巴西基酸(1.54g,6.31mmol)和4-二甲基氨基吡啶(dmap,1.54g,12.62mmol)溶于水(10cm3)中,并将样品置于显微镜上。观察到直径~3μm的几乎都为单分散的球形实体(图1)。
将聚ε-赖氨酸(pek)(2g,12.04mmolnh2)溶于水(10cm3)中,并加入到上述巴西基酸/dmap微球的溶液中。将混合物通过0.45μm膜过滤并将样品置于显微镜上。直径~3μm的微球仍然存在。将该溶液用水稀释至100cm3。将n-(3-二甲基氨基丙基)-n'-乙基碳二亚胺盐酸盐(edcl)(4.6g,2.4mmol)和honsu(1.38g,1.2mmol)溶于水(10cm3)中,并加入到上述溶液中。使交联反应进行过夜,通过切向流过滤(tff)进行洗涤并通过冷冻干燥回收所得颗粒(产量2.35g)。图2显示所得微球的扫描电子显微照片。
实施例6-含有原卟啉ix,血红素b的交联的自组装微粒的制备
将巴西基酸(0.734g,3.3mmol)和4-二甲基氨基吡啶(dmap,0.734g,6.6mmol)溶于水(10cm3)中,并将样品置于显微镜上。观察到直径~3μm的几乎都为单分散的球形实体(图1)。
将聚ε-赖氨酸(pek)(1g,6.02mmol的nh2)溶解在水(10cm3)中,并加入到上述巴西基酸/dmap微球的溶液中。将混合物通过0.45μm膜过滤并将样品置于显微镜上。直径~3μm的微球仍然存在。将该溶液用血红素b(50cm3)的饱和溶液稀释。将n-(3-二甲基氨基丙基)-n'-乙基碳二亚胺盐酸盐(edcl)(2.3g,1.2mmol)和honsu(0.7g,0.6mmol)溶于水(5cm3)中,并加入到上述溶液中。使交联反应进行过夜,通过切向流过滤(tff)进行洗涤并通过冷冻干燥回收所得颗粒(产量0.93g)。
实施例7-交联的自组装微粒的制备
将癸二酸(0.619g,6.12mmol)和nmm(0.62g,6.12mmol)溶于水(10cm3)中,并将样品置于显微镜上。观察到直径~2.5μm的几乎都为单分散的球形实体。
将聚ε-赖氨酸(pek)(1g,5.83mmolnh2)溶于水(10cm3)中,并加入到上述癸二酸/nmm微球的溶液中。将混合物通过0.45μm膜过滤并将样品置于显微镜上。直径~2.5μm的微球仍然存在。将该溶液用水稀释至50cm3。将edcl(2.24g,11.7mmol)和honsu(2.0g,17.4mmol)溶于水(10cm3)中并加入到上述溶液中。使交联反应进行过夜,通过tff进行洗涤并通过冻干回收所得颗粒。
实施例8-交联的自组装微粒的制备
将癸二酸(5.06g,25mmol)和咪唑(3.4g,50mmol)溶于水(50cm3)中,并将样品置于显微镜上。观察到直径~2.5μm的几乎都为单分散的球形实体。
将聚ε-赖氨酸(pek)(8.576g,50mmolnh2)溶于水(50cm3)中,并加入到上述癸二酸/咪唑微球的溶液中。将混合物通过0.45μm膜过滤并将样品置于显微镜上。直径~2.5μm的微球仍然存在(图3)。将该溶液用水稀释至500cm3。将edcl(4.8g,25mmol)溶于水(20cm3)中并加入到上述溶液中。使交联反应进行1小时,然后再加入25mmoledcl,放置过夜。通过倾析用水进行洗涤并通过冷冻干燥回收所得颗粒(图4)。
实施例9-交联的自组装微粒的制备
将癸二酸(5g,24.7mmol)和(3-氨基丙基)三甲氧基硅烷(8.42g,46.9mmol)溶于水(50cm3)中,并将样品置于显微镜上。观察到直径~2.5μm的几乎都为单分散的球形实体。
将混合物放置过夜,然后用浓盐酸酸化。加入盐酸使得在颗粒内形成二氧化硅,产生癸二酸/二氧化硅复合物。
实施例10-交联的自组装微粒的制备
将癸二酸(5g,24.7mmol)和n-[3-(三甲氧基甲硅烷基)丙基]乙二胺(5.77g,51.9mmol胺)溶于水(50cm3)中,并将样品置于显微镜上。观察到直径~2.5μm的几乎都为单分散的球形实体。
将该溶液用水稀释至500cm3。将edcl(20g,104mmol)溶于水(100cm3)中并加入到上述溶液中。将混合物放置过夜,然后用浓盐酸酸化。加入盐酸使得在颗粒内形成二氧化硅,产生癸二酸/二氧化硅复合物。
实施例11-交联的自组装微粒的制备
将癸二酸(5g,24.7mmol)和n1-(3-三甲氧基甲硅烷基丙基)二亚乙基三胺(4.37g,46.9mmol胺)溶于水(50cm3)中,并将样品置于显微镜上。观察到直径~2.5μm的几乎都为单分散的球形实体。
将该溶液用水稀释至500cm3。将edcl(20g,104mmol)溶于水(100cm3)中并加入到上述溶液中。将混合物放置过夜,然后用浓盐酸酸化。加入盐酸使得在颗粒内形成二氧化硅,产生癸二酸/二氧化硅复合物。
实施例12-自组装大孔交联的片材的制备
将癸二酸(0.619g,6.12mmol)和nmm(0.62g,6.12mmol)溶于水(10cm3)中,并将样品置于显微镜上。观察到直径~2.5μm的几乎都为单分散的球形实体。
将聚ε-赖氨酸(pek)(1g,5.83mmolnh2)溶于水(10cm3)中,并加入到上述癸二酸/nmm微球的溶液中。将混合物通过0.45μm膜过滤并将样品置于显微镜上。直径~2.5μm的微球仍然存在。将edcl(2.24g,11.7mmol)和honsu(2.0g,17.4mmol)溶于水(10cm3)中并加入到上述溶液中。使交联反应进行过夜,用水进行洗涤并通过冻干法干燥得到的片材。图5所示的sem清楚地显示了形成的大孔聚合物的融合微球结构。
实施例13-自组装大孔交联的片材的制备
将(12-膦基十二烷基)膦酸(330mg,1mmol)和nmm(404mg,4mmol)溶解于水。置于显微镜上的样品证实存在几乎都为单分散的微球。将pek(343mg,2mmolnh2)溶于水(10cm3)并加入到上述制备的双膦酸溶液中。此阶段仍然存在微球。加入溶于水(10cm3)中的edcl(1.15g,6mmol),并将混合物立即倒入托盘中。在这个阶段还是仍然存在微球。用水彻底洗涤在~2h后形成的片材。最终的片材具有橡胶质地。
实施例14:骨细胞培养(3d细胞培养的实例)
在本实施例中,制造了实施例12中制造的自组装大孔片材。测试了另一种包含实施例12的片材以及相对于支架为约10%重量的羟基磷灰石纳米颗粒的产品(所述羟基磷灰石纳米颗粒可从sigmaaldrich获得,目录号702153)和细胞相容性。
成骨细胞骨细胞的9天生长曲线表明,与组织培养塑料对照相比,在培养期的早期阶段,细胞活力无显着差异。在羧基或羟基磷灰石涂覆的支架上培养的成骨细胞之间的细胞活力没有显着差异。与3d支架的细胞相互作用在×40放大倍率下变得更加明显,并且肌动蛋白丝染色突出显示了细胞锚定到3d支架的附着点,如图7所示。
图7显示在羧基官能化的和羟基磷灰石涂覆的3d支架上培养的成骨细胞,a+b)培养48小时,×10放大倍率,c+d)培养48小时,×40放大倍率,e+f)培养7天,×10放大倍率,g+h)培养7天,×20放大倍率。
在剩下的培养时期,成骨细胞继续存活并与支架相互作用。这些结果表明具有/不具有羟基磷灰石涂层的3d支架支持成骨细胞生长。
成骨细胞增殖
将成骨细胞以1×105个细胞的密度接种于24孔插入物上,并在37℃和5%co2的标准组织培养条件下培养。使用含有10%fcs与两性菌素b和pen/strep补充剂的dulbecco's改良的eagle培养基(dmem)(高葡萄糖+2mm谷氨酰胺)。用fitc缀合的phalloidin(鬼笔环肽)对细胞f-肌动蛋白进行染色,然后用核染色剂hoechst33342进行复染。使用nikoneclipseti-e相差显微镜(nikon,tokyo,japan)在24小时、48小时和7天的时间点拍摄图像(图6)。图6:在9天的培养期间对羧基和羟基磷灰石涂覆的3d支架上培养的成骨细胞的代谢活性测定(cck-8)。
细胞增殖测定
在9天的细胞培养期间使用cck-8测定试剂盒(dojindolaboratories,kumamoto,japan)在不同时间段监测细胞增殖。根据制造商的指导方案建立标准曲线以确定细胞数量。在以5×104个细胞的初始密度向支架上接种之后在标准培养条件下孵育细胞。在每个时间点将cck-8溶液(50mm3)加入每个孔中的培养基(500mm3)。然后将细胞在标准培养条件下孵育2小时。将来自每个孔的等分试样(3×100mm3)溶液移至96孔板中标记的孔中。也使用合适的对照。使用fluostaroptima平板读数器(bmglabtech,ortenberg,germany)在485nm读取吸光度,600nm为背景读数,记录结果。
实施例15-杀生物剂制品
出于磨损的原因,目前用于个人护理、化妆品、家庭护理和一般消毒的杀生物剂受到它们保持与待处理表面接触的时间的限制。例如,用于在医院内消毒的表面喷雾剂类型具有有限的活性寿命,并因此降低了对例如mrsa、铜绿假单胞菌(p.auregenosa)和艰难梭菌(c.difficile)的医院感染的活性。此外,一些表面喷雾剂含有有机溶剂如异丙醇或非生物可降解的组分如硅油,以减少杀生物剂的磨损去除。
阳离子和两性杀生物剂如季铵化合物通过溶解细胞膜,导致细胞裂解和死亡而对病原体起作用。商业上有许多杀生物剂用于消毒,包括氯己定、苯扎氯铵、氯咪巴唑、二癸基二甲基氯化铵、十二烷基二亚丙基三胺,它们是阳离子化合物。另外,一些杀生物剂是聚合的阳离子化合物如聚(二烯丙基二甲基氯化铵)。这些化合物可以使用本文所述的技术容易地配制成球形微粒,这将允许在表面、皮肤和头发上减少磨损去除;潜在地允许杀菌剂的受控释放。此外,在同一微粒中含有多个阳离子化合物的杀生物剂是可能的,并且可以提供可以定制并靶向其中感染源得到良好定义的具体应用的制剂。
所生产的样品如下:
聚(二烯丙基二甲基氯化铵)(pdac)spherisomes
将pdac(1.615g,10mmol)溶于水(50cm3)中,并加入naoh(0.4g,10mmol)。向该溶液中加入巴西基酸(1.22g,5mmol),并使其溶解过夜。这看上去是清澈的溶液,但是在显微镜下观察时,确认是~3μm微粒和pdac新制剂的悬浮液,并且结果如图8所示,显示了pdac-巴西基酸微粒制剂。
二癸基二甲基氯化铵(ddac)
将ddac(9.04cm3的40%w/v溶液,10mmol)用水稀释至50cm3,并加入naoh(0.4g,10mmol)。向该溶液中加入巴西基酸(1.22g,5mmol),并使其溶解过夜。这看上去是不透明的溶液,但在显微镜下观察时,确认是~3μm微粒和ddac的新制剂的悬浮液。
十二烷基二亚丙基三胺(ddpt)
将ddpt(9.97cm3的30%w/v溶液,10mmol)用水稀释至50cm3,并向该溶液中加入巴西基酸(3.66g,15mmol),使其溶解过夜。这看上去是清澈的溶液,但在显微镜下观察时,确认是~3μm微粒和ddpt新制剂的悬浮液。
实施例16-抗微生物伤口敷料
通过双羧基脂肪酸微粒的碰撞形成的多孔聚合物的亲水特性对于用于吸收性伤口敷料是有利的。当双羧基脂肪酸与聚ε-赖氨酸组合并交联而形成这种多孔基质时,在需要时可以保持和增强伤口敷料组分的天然抗微生物活性。在阳离子形式中,聚-ε-赖氨酸超过脂肪酸,已证明材料保留了新型抗微生物伤口敷料的食品防腐剂的特性。材料的多孔性质将允许改进的皮肤修复,因为3d支架组合阳离子特质能够破坏微生物生物膜。
使用混合的物种cdc反应器模型评估阳离子伤口敷料的抗生物膜能力。这些实验中使用了实施例13的产品。
如下所示制备两种混合的物种生物膜,并针对pbs和对照阴离子敷料进行测试。
多物种生物膜1
金黄色葡萄球菌(staphylococcusaureus)nctc8325
铜绿假单胞菌(pseudomonasaeruginosa)ncimb10434
鲍曼不动杆菌(acinetobacterbaumannii)atcc19606
表皮葡萄球菌(staphylococcusepidermidis)
多物种生物膜2
金黄色葡萄球菌nctc8325
mrsa
vre粪肠球菌(faecalis)nctc12201
白色念珠菌(candidaalbicans)atccmya-2876sc5313
大肠杆菌(escherichiacoli)nctc129233/6页,dot202(03)
混合接种物1的制备
使用无菌拭子从适当的琼脂平板上收获金黄色葡萄球菌、铜绿假单胞菌、鲍曼不动杆菌和表皮葡萄球菌的24小时培养物,并将其悬浮在20cm3的胰蛋白胨大豆肉汤(tsb)中。将混合的物种悬浮液在tsb中稀释,而得到107±5×106cfuml-1的总浓度,并用作cdc反应器的接种物。将cdc反应器在37℃以50rpm摇动孵育72小时,以促进生物膜生长。
混合接种物2的制备
使用无菌拭子从适当的琼脂平板上收获金黄色葡萄球菌、耐甲氧西林金黄色葡萄球菌(methicillin-resistantstaphylococcusaureus)、耐万古霉素肠球菌(vancomycin-resistantenterococcus)、白色念珠菌和大肠杆菌的24小时培养物,并悬浮于20cm3的tsb中。将混合的悬浮液在tsb中稀释,而得到107±5×106cfuml-1的总浓度,并用作cdc反应器的接种物。将cdc反应器在37℃以50rpm摇动孵育72小时,以促进生物膜生长。
生物膜处理
孵育后,从cdc反应器中取出试样,并在无菌磷酸盐缓冲盐水(pbs)中洗涤3次,以除去浮游生物细胞。然后通过将试样放置在两块伤口敷料材料中间来处理洗涤后的试样。在测试前通过向每块敷料添加400mm3pbs+1%tsb以激活敷料。将对照试样浸没在1cm3的pbs+1%tsb中。所有样品一式三份进行测试。在24小时处理期后,将试样放置在1cm3的pbs中并超声处理15分钟,以回收附着于试样的任何活微生物。使用连续稀释液和平板涂布法定量回收的微生物。
混合的接种物1
如图9所示,用对照敷料(a)进行处理后,细菌回收情况与仅pbs处理对照相似。从用阳离子敷料(b)处理的试样中没有回收到活的生物体。这表示与pbs处理的对照相比,大于5log的减少。处理后存活的生物体主要是铜绿假单胞菌(图10)。
混合的接种物2
与pbs处理的对照相比,对照敷料(a)的处理导致回收的活细菌数量1.27log的减少。从阳离子敷料(b)处理的试样中没有回收到活的生物体。这表示与pbs处理的对照相比,大于7log的减少(图11)。存活的生物体是混合的物种(图12)。
- 还没有人留言评论。精彩留言会获得点赞!