一种2‑(4‑溴‑1‑甲基‑1H‑吡唑‑5‑基)乙胺的制备方法与流程
本发明涉及医药中间体领域,具体涉及一种2-(4-溴-1-甲基-1h-吡唑-5-基)乙胺的制备方法。
背景技术:
吡唑及其衍生物是一类重要的化合物,具有较强的生物活性,广泛应用于医药、农药等领域,因此吡唑衍生物的合成受到了广泛关注,尤其是在医药中间体之中被广泛应用。cn103891743b中给出了一种含吡唑萘菌胺和吡唑醚菌酯的杀菌组合物,cn102702212b中给出了一种n-(2-取代酰基)吡唑鱼藤酚,cn102267986b中给出了一种吡唑双酰胺类化合物及其合成方法与应用,这都是吡唑类衍生物应用的示例,但是本申请提及的2-(4-溴-1-甲基-1h-吡唑-5-基)乙胺鲜少有作为医药中间体的应用。由于该分子的特性,该方法不能推广到其他类似结构的合成中。
技术实现要素:
本发明主要是提供一种2-(4-溴-1-甲基-1h-吡唑-5-基)乙胺及其制备方法。该方法步骤清晰,浪费少,产率较高,节省原料,整体较为经济。
本发明的上述技术问题主要是通过下述技术方案得以解决的:
一种医药中间体2-(4-溴-1-甲基-1h-吡唑-5-基)乙胺,其特征在于,具有如下所述结构:
一种所述的2-(4-溴-1-甲基-1h-吡唑-5-基)乙胺的制备方法,其特征在于包括如下步骤:
s1、在氮气气氛保护下,将90-120g的1-甲基吡唑加入到2l四氢呋喃中,搅拌溶解至均一状态,然后冷却到零下60~零下80摄氏度,再滴加2.5m浓度的正丁基锂600ml,保温在零下60摄氏度以下,在正丁基锂滴加完毕后,再保温0.4-0.7h,冷却至零下65至零下80摄氏度,滴加100mln,n-二甲基甲酰胺,滴完n,n-二甲基甲酰胺后室温过夜,整体冷却至0摄氏度,后滴加300-500ml去离子水淬灭。优选地,所述氮气气氛保护下,是将氮气深入到液面下持续通入,所述冷却和保温均要在保证温度稳定15min中在进行下一步操作。
s2、每次用500ml乙酸乙酯萃取,重复2-4次,分离出有机相,每次用500ml饱和食盐水洗涤,重复洗2-4次,用50g-150g无水硫酸钠干燥1-2h,抽滤,再进行减压旋蒸,得s2步骤产品1-甲基-1h-吡唑-5-甲醛;优选地,所述分离出有机相要在液体稳定3min以上再尽心,无水硫酸钠使用之前过200目筛,所述减压旋蒸的减压幅度为0.05-0.15个大气压。
s3、将s2步骤产品1-甲基-1h-吡唑-5-甲醛取130-170g加入到1100-1300ml的吡啶中,再加入10-14ml哌啶,140-150g丙二酸,然后升温至70-90摄氏度,反应0.5-1.5h,再保持温度回流3h,在经过冷却步骤,在减压条件下将吡啶全部蒸发掉,加入200-400ml乙醇,析出固体,将析出的固体进行抽滤和烘干,得到s3步骤产物3-(1-甲基-1h-吡唑-5-基)丙烯酸,常温下为固体;优选地,所述哌啶,丙二酸的加入过程中持续搅拌,所述保持温度回流是保持在80摄氏度左右,所述减压的减压幅度为0.05-0.15个大气压,所述析出固体的过程要等待固体停止析出后10min再操作。
s4、取s3步骤产物3-(1-甲基-1h-吡唑-5-基)丙烯酸30-36g加入到500-700ml甲醇中,然后加入20-30g的钯碳,然后通入氢气,进行加氢反应2-4小时,反应结束后,通过抽滤手段除去全部钯碳,将甲醇全部蒸发去除,得到s4步骤产品3-(1-甲基-1h-吡唑)丙酸;优选地,所述通入氢气是将氮气深入到液面下持续通入,抽滤手段去除全部钯碳,为了保证去除完全可重复进行1-2次,甲醇蒸发过程仅稍稍加热,保证仅蒸发甲醇。
s5、将s4步骤产品3-(1-甲基-1h-吡唑)丙酸15-25g溶于200-400ml乙酸中,然后滴入1.25eq溴素,后加热至60-80摄氏度,反应3-5h,再加入饱和亚硫酸钠溶液40-60ml,再进行旋干蒸发得到油状物,放置一夜成s5步骤产物固体3-(4-溴-1-甲基-1h-吡唑-5-基)丙酸;优选地,所述放置一夜时额外加装含有干燥物质的干燥器,以保证干燥效果。
s6、取65-75ml氯化亚砜,放入容器内,降温到0摄氏度,然后加入s5步骤产物3-(4-溴-1-甲基-1h-吡唑-5-基)丙酸20-25g,加完后保温10-20min,后升温至35-45摄氏度反应1.5-2.5h,蒸发全部氯化亚砜,加入150-250ml四氢呋喃,降温至0摄氏度以下,滴入到有250-300ml浓氨水的四口瓶中,滴完搅拌10-20min,自然缓慢升至室温,反应完全后,以旋干挥发形式蒸发掉所有氨水,用200ml甲醇溶出产物,再次旋干后,用100-200目硅胶纯化,利用乙酸乙酯做为洗脱剂,得到s6步骤产物3-(4-溴-1-甲基-1h-吡唑-5-基)丙酰胺;优选地,所述降温至0摄氏度以下以在外包裹冰水混合物并持续加冰以保证温度。
s7、取18-22gnaoh加入到150-200ml水中,然后降温至0摄氏度,然后加入s6步骤产物3-(4-溴-1-甲基-1h-吡唑)丙酰胺10-15g,后滴入1.2eq溴素,然后搅拌5-10min,撤去冰浴,然后加热至55-65℃,保持此温度过夜,加热过程中固体逐渐溶解,反应液冷却,用300ml乙酸乙酯萃取,用适量无水硫酸钠干燥,旋干得产品2-(4-溴-1-甲基-1h-吡唑-5-基)乙胺。优选地,保持此温度过夜,指的是至少反应时间在10h以上。前述所有试剂均为化学纯以上纯度,或者均为优级纯。
与现有技术相比,本发明的优点在于:是以1-甲基吡唑为原料,反应形成1-甲基-1h-吡唑-5-甲醛;然后加入哌啶和吡啶得到固体3-(1-甲基-1h-吡唑-5-基)丙烯酸;进一步在甲醇溶液中加入钯碳,通氢气,得到3-(1-甲基-1h-吡唑)丙酸,再进一步,在溴素和饱和亚硫酸钠溶液的作用下反应,经旋干得到固体3-(4-溴-1-甲基-1h-吡唑-5-基)丙酸,再依次经氯化亚砜、四氢呋喃加入反应,以及甲醇溶出、硅胶纯化、乙酸乙酯洗脱,得到3-(4-溴-1-甲基-1h-吡唑-5-基)丙酰胺,最后加入naoh和溴素,在冰浴的环境下得到2-(4-溴-1-甲基-1h-吡唑-5-基)乙胺。本文巧妙地利用了钯碳的作用,定向选择性地将中间的双键加氢,又通过定量naoh和溴素的方式去掉了羰基,氨基仍得以保留,体现了极强的发明构思和创造性,在现有技术中并无较为类似的公开信息可供借鉴,本发明方案具有独创性。该方法具有步骤清晰,浪费少,产率较高,节省原料且易于操作的优点。
附图说明
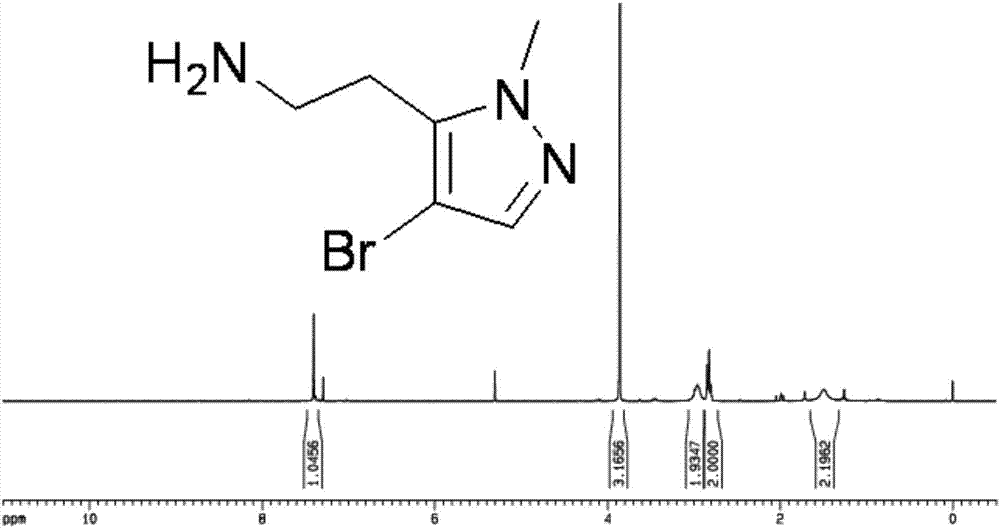
图1是本发明的2-(4-溴-1-甲基-1h-吡唑-5-基)乙胺的hnmr谱图;
图2是本发明的2-(4-溴-1-甲基-1h-吡唑-5-基)乙胺的hplc谱图。
具体实施方式
下面结合附图对本发明的优选实施例进行详细阐述,以使本发明的优点和特征能更易于被本领域技术人员理解,从而对本发明的保护范围做出更为清楚明确的界定。本发明可以以许多不同的形式实施,而不应该被理解为限于在此阐述的实施例。相反,提供这些实施例,使得本公开将是彻底的和完整的,并且将把本发明的构思充分传达给本领域技术人员,本发明将仅由权利要求来限定。本发明提供的2-(4-溴-1-甲基-1h-吡唑-5-基)乙胺的制备方法,其工艺路线如下:
其合成方法可以简述如下:是以1-甲基吡唑为原料,反应形成1-甲基-1h-吡唑-5-甲醛;然后加入哌啶和吡啶得到固体3-(1-甲基-1h-吡唑-5-基)丙烯酸;进一步在甲醇溶液中加入钯碳,通氢气,得到3-(1-甲基-1h-吡唑)丙酸,再进一步,在溴素和饱和亚硫酸钠溶液的作用下反应,经旋干得到固体3-(4-溴-1-甲基-1h-吡唑-5-基)丙酸,再依次经氯化亚砜、四氢呋喃加入反应,以及甲醇溶出、硅胶纯化、乙酸乙酯洗脱,得到3-(4-溴-1-甲基-1h-吡唑-5-基)丙酰胺,最后加入naoh和溴素,在冰浴的环境下得到2-(4-溴-1-甲基-1h-吡唑-5-基)乙胺。
实施例1
s1、在氮气气氛保护下,将100g的1-甲基吡唑加入到2l四氢呋喃中,搅拌溶解至均一状态,然后冷却到零下60度,再滴加2.5m浓度的正丁基锂600ml,保温在零下60摄氏度以下,在正丁基锂滴加完毕后,再保温0.4h,冷却至零下65摄氏度,滴加100mln,n-二甲基甲酰胺,滴完n,n-二甲基甲酰胺后室温过夜,整体冷却至0摄氏度,后滴加300ml去离子水淬灭。
s2、每次用500ml乙酸乙酯萃取,重复2次,分离出有机相,每次用500ml饱和食盐水洗涤,重复洗2次,用50g无水硫酸钠干燥1h,抽滤,再进行减压旋蒸,得s2步骤产品1-甲基-1h-吡唑-5-甲醛。
s3、将s2步骤产品1-甲基-1h-吡唑-5-甲醛取130g加入到1100ml的吡啶中,再加入10ml哌啶,140g丙二酸,然后升温至70摄氏度,反应0.5h,再保持温度回流3h,在经过冷却步骤,在减压条件下将吡啶全部蒸发掉,加入200ml乙醇,析出固体,将析出的固体进行抽滤和烘干,得到s3步骤产物3-(1-甲基-1h-吡唑-5-基)丙烯酸,常温下为固体。
s4、取s3步骤产物3-(1-甲基-1h-吡唑-5-基)丙烯酸30g加入到500ml甲醇中,然后加入20g的钯碳,然后通入氢气,进行加氢反应2小时,反应结束后,通过抽滤手段除去全部钯碳,将甲醇全部蒸发去除,得到s4步骤产品3-(1-甲基-1h-吡唑)丙酸。
s5、将s4步骤产品3-(1-甲基-1h-吡唑)丙酸15g溶于200ml乙酸中,然后滴入1.25eq溴素,后加热至60摄氏度,反应3h,再加入饱和亚硫酸钠溶液40ml,再进行旋干蒸发得到油状物,放置一夜成s5步骤产物固体3-(4-溴-1-甲基-1h-吡唑-5-基)丙酸。
s6、取65ml氯化亚砜,放入容器内,降温到0摄氏度,然后加入s5步骤产物3-(4-溴-1-甲基-1h-吡唑-5-基)丙酸20g,加完后保温10min,后升温至35摄氏度反应1.5h,蒸发全部氯化亚砜,加入150ml四氢呋喃,降温至0摄氏度以下,滴入到有250ml浓氨水的四口瓶中,滴完搅拌10-20min,自然缓慢升至室温,反应完全后,以旋干挥发形式蒸发掉所有氨水,用200ml甲醇溶出产物,再次旋干后,用100-200目硅胶纯化,利用乙酸乙酯做为洗脱剂,得到s6步骤产物3-(4-溴-1-甲基-1h-吡唑-5-基)丙酰胺。
s7、取18gnaoh加入到150ml水中,然后降温至0摄氏度,然后加入s6步骤产物3-(4-溴-1-甲基-1h-吡唑)丙酰胺10g,后滴入1.2eq溴素,然后搅拌5min,撤去冰浴,然后加热至55℃,保持此温度过夜,加热过程中固体逐渐溶解,反应液冷却,用300ml乙酸乙酯萃取,用适量无水硫酸钠干燥,旋干得产品2-(4-溴-1-甲基-1h-吡唑-5-基)乙胺。
实施例2
s1、在氮气气氛保护下,将110g的1-甲基吡唑加入到2l四氢呋喃中,搅拌溶解至均一状态,然后冷却到零下70摄氏度,再滴加2.5m浓度的正丁基锂600ml,保温在零下60摄氏度以下,在正丁基锂滴加完毕后,再保温0.5h,冷却至零下70摄氏度,滴加100mln,n-二甲基甲酰胺,滴完n,n-二甲基甲酰胺后室温过夜,整体冷却至0摄氏度,后滴加400ml去离子水淬灭。
s2、每次用500ml乙酸乙酯萃取,重复3次,分离出有机相,每次用500ml饱和食盐水洗涤,重复洗3次,用100g无水硫酸钠干燥1.5h,抽滤,再进行减压旋蒸,得s2步骤产品1-甲基-1h-吡唑-5-甲醛。
s3、将s2步骤产品1-甲基-1h-吡唑-5-甲醛取150g加入到1200ml的吡啶中,再加入12ml哌啶,143g丙二酸,然后升温至80摄氏度,反应1h,再保持温度回流3h,在经过冷却步骤,在减压条件下将吡啶全部蒸发掉,加入300ml乙醇,析出固体,将析出的固体进行抽滤和烘干,得到s3步骤产物3-(1-甲基-1h-吡唑-5-基)丙烯酸,常温下为固体。
s4、取s3步骤产物3-(1-甲基-1h-吡唑-5-基)丙烯酸33g加入到600ml甲醇中,然后加入25g的钯碳,然后通入氢气,进行加氢反应3小时,反应结束后,通过抽滤手段除去全部钯碳,将甲醇全部蒸发去除,得到s4步骤产品3-(1-甲基-1h-吡唑)丙酸。
s5、将s4步骤产品3-(1-甲基-1h-吡唑)丙酸20g溶于300ml乙酸中,然后滴入1.25eq溴素,后加热至70摄氏度,反应4h,再加入饱和亚硫酸钠溶液50ml,再进行旋干蒸发得到油状物,放置一夜成s5步骤产物固体3-(4-溴-1-甲基-1h-吡唑-5-基)丙酸。
s6、取70ml氯化亚砜,放入容器内,降温到0摄氏度,然后加入s5步骤产物3-(4-溴-1-甲基-1h-吡唑-5-基)丙酸23g,加完后保温15min,后升温至40摄氏度反应2h,蒸发全部氯化亚砜,加入200ml四氢呋喃,降温至0摄氏度以下,滴入到有280ml浓氨水的四口瓶中,滴完搅拌15min,自然缓慢升至室温,反应完全后,以旋干挥发形式蒸发掉所有氨水,用200ml甲醇溶出产物,再次旋干后,用150目硅胶纯化,利用乙酸乙酯做为洗脱剂,得到s6步骤产物3-(4-溴-1-甲基-1h-吡唑-5-基)丙酰胺。
s7、取20gnaoh加入到180ml水中,然后降温至0摄氏度,然后加入s6步骤产物3-(4-溴-1-甲基-1h-吡唑)丙酰胺12g,后滴入1.2eq溴素,然后搅拌8min,撤去冰浴,然后加热至60℃,保持此温度过夜,加热过程中固体逐渐溶解,反应液冷却,用300ml乙酸乙酯萃取,用适量无水硫酸钠干燥,旋干得产品2-(4-溴-1-甲基-1h-吡唑-5-基)乙胺。
实施例3
s1、在氮气气氛保护下,将120g的1-甲基吡唑加入到2l四氢呋喃中,搅拌溶解至均一状态,然后冷却到零下80摄氏度,再滴加2.5m浓度的正丁基锂600ml,保温在零下60摄氏度以下,在正丁基锂滴加完毕后,再保温0.7h,冷却至零下80摄氏度,滴加100mln,n-二甲基甲酰胺,滴完n,n-二甲基甲酰胺后室温过夜,整体冷却至0摄氏度,后滴加500ml去离子水淬灭。
s2、每次用500ml乙酸乙酯萃取,重复4次,分离出有机相,每次用500ml饱和食盐水洗涤,重复洗4次,用150g无水硫酸钠干燥2h,抽滤,再进行减压旋蒸,得s2步骤产品1-甲基-1h-吡唑-5-甲醛。
s3、将s2步骤产品1-甲基-1h-吡唑-5-甲醛取170g加入到1300ml的吡啶中,再加入14ml哌啶,150g丙二酸,然后升温至90摄氏度,反应1.5h,再保持温度回流3h,在经过冷却步骤,在减压条件下将吡啶全部蒸发掉,加入400ml乙醇,析出固体,将析出的固体进行抽滤和烘干,得到s3步骤产物3-(1-甲基-1h-吡唑-5-基)丙烯酸,常温下为固体。
s4、取s3步骤产物3-(1-甲基-1h-吡唑-5-基)丙烯酸36g加入到700ml甲醇中,然后加入30g的钯碳,然后通入氢气,进行加氢反应4小时,反应结束后,通过抽滤手段除去全部钯碳,将甲醇全部蒸发去除,得到s4步骤产品3-(1-甲基-1h-吡唑)丙酸。
s5、将s4步骤产品3-(1-甲基-1h-吡唑)丙酸25g溶于400ml乙酸中,然后滴入1.25eq溴素,后加热至80摄氏度,反应5h,再加入饱和亚硫酸钠溶液60ml,再进行旋干蒸发得到油状物,放置一夜成s5步骤产物固体3-(4-溴-1-甲基-1h-吡唑-5-基)丙酸。
s6、取75ml氯化亚砜,放入容器内,降温到0摄氏度,然后加入s5步骤产物3-(4-溴-1-甲基-1h-吡唑-5-基)丙酸25g,加完后保温20min,后升温至45摄氏度反应2.5h,蒸发全部氯化亚砜,加入250ml四氢呋喃,降温至0摄氏度以下,滴入到有300ml浓氨水的四口瓶中,滴完搅拌20min,自然缓慢升至室温,反应完全后,以旋干挥发形式蒸发掉所有氨水,用200ml甲醇溶出产物,再次旋干后,用100-200目硅胶纯化,利用乙酸乙酯做为洗脱剂,得到s6步骤产物3-(4-溴-1-甲基-1h-吡唑-5-基)丙酰胺。
s7、取22gnaoh加入到200ml水中,然后降温至0摄氏度,然后加入s6步骤产物3-(4-溴-1-甲基-1h-吡唑)丙酰胺15g,后滴入1.2eq溴素,然后搅拌10min,撤去冰浴,然后加热至65℃,保持此温度过夜,加热过程中固体逐渐溶解,反应液冷却,用300ml乙酸乙酯萃取,用适量无水硫酸钠干燥,旋干得产品2-(4-溴-1-甲基-1h-吡唑-5-基)乙胺。
以上所述,仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,任何不经过创造性劳动想到的变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应该以权利要求书所限定的保护范围为准。
- 还没有人留言评论。精彩留言会获得点赞!