一种16α,16β,17β‑三氘‑1,4‑雄烯二酮的合成方法与流程
本发明涉及有机合成领域,特别涉及一种内标物16α,16β,17β-三氘-1,4-雄烯二酮的合成方法。
背景技术:
随着色谱检测的普及,稳定同位素标记的标准品已成为最理想的定量内标物,以稳定同位素(如2h、13c、15n、18o)作为内标,不仅提高了实验数据的可靠性,而且还提高了测试灵敏度。16α,16β,17β-三氘-1,4-雄烯二酮,俗称17β-勃地酮-d3,采用2h为稳定同位素,是一种极其稳定、信号强又不会产生干扰的内标物,常用于兽药残留的检测,在待分析样品中加入17β-勃地酮-d3作为内标物进行检测即可。目前,17β-勃地酮-d3的合成路线复杂,氘代率低,合成产物复杂,后处理过程繁琐,导致生产成本很高,我国对此类标准品基本只能依靠国外进口。
技术实现要素:
为了解决上述现有技术方案中的不足,本发明提供了一种反应路线简单、反应条件温和、产物纯度高、氘代率高、成本低的16α,16β,17β-三氘-1,4-雄烯二酮的合成方法。
本发明的目的是通过以下技术方案实现的:
一种16α,16β,17β-三氘-1,4-雄烯二酮的合成方法,所述合成方法包括以下步骤:
(a1)1,4-雄烯二酮与氘代水在第一溶剂中混合,并在弱碱性盐的催化作用下加热回流,1,4-雄烯二酮与氘代水发生氢氘交换;
(a2)萃取回流反应液,浓缩萃取的有机相后得到中间产物16α,16β–二氘-1,4-雄烯二酮;
(a3)所述中间产物与硼氘化钠在第二溶剂中混合,并在冰浴条件下进行选择性还原;
(a4)酸化还原反应液使其ph≤2;
(a5)萃取酸化后的还原反应液,采用柱层析法分离有机相,获得目标产物16α,16β,17β-三氘-1,4-雄烯二酮。
根据上述的合成方法,可选地,1,4-雄烯二酮与氘代水的物质的量比为1:100~1:500。
根据上述的合成方法,可选地,所述第一溶剂与氘代水的体积比为1:2~1:4。
根据上述的合成方法,优选地,(a1)步骤中,加热回流时间为24~30小时。
根据上述的合成方法,优选地,所述第一溶剂为与水互溶的有机溶剂。
根据上述的合成方法,优选地,所述第一溶剂为四氢呋喃。
根据上述的合成方法,优选地,所述弱碱性盐为碳酸钾。
根据上述的合成方法,可选地,所述中间产物与硼氘化钠的物质的量之比为1:1~1:1.2。
根据上述的合成方法,可选地,冰浴温度为-10℃~0℃,冰浴反应时间为20-60分钟。
根据上述的合成方法,优选地,所述第二溶剂为氘代甲醇。
根据上述的合成方法,优选地,(a2)、(a5)步骤中,采用乙酸乙酯进行萃取。
与现有技术相比,本发明具有的有益效果为:
1、本发明创造性地提出了16α,16β,17β-三氘-1,4-雄烯二酮的一种合成方法,即采用1,4-雄烯二酮为原料,先与氘代水进行氢氘交换,再通过硼氘化钠进行选择性还原得到目标产物,合成路线简单,反应原料廉价易得,成本低。
2、本发明操作简单,反应条件温和,产率稳定。
3、本发明的合成方法获得的产物纯度在99%以上,氘代率在98.5%以上。
附图说明
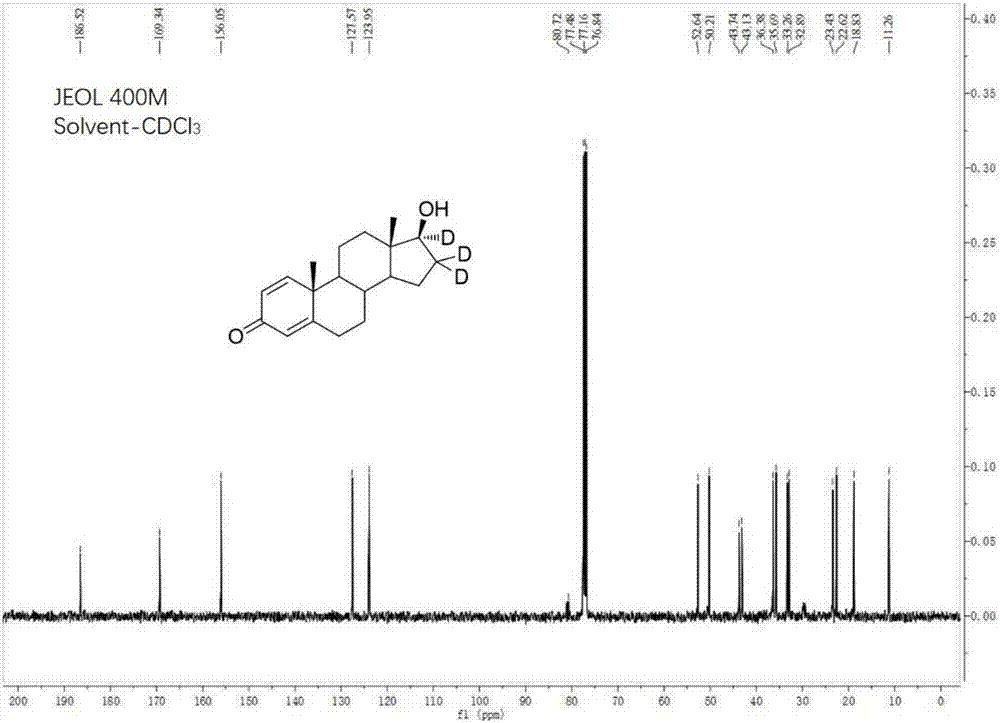
图1为16α,16β,17β-三氘-1,4-雄烯二酮的1hnmr图谱;
图2为16α,16β,17β-三氘-1,4-雄烯二酮的13cnmr图谱。
具体实施方式
图1-2和以下说明描述了本发明的可选实施方式以教导本领域技术人员如何实施和再现本发明。为了教导本发明技术方案,已简化或省略了一些常规方面。本领域技术人员应该理解源自这些实施方式的变型或替换将在本发明的范围内。本领域技术人员应该理解下述特征能够以各种方式组合以形成本发明的多个变型。由此,本发明并不局限于下述可实施方式,而仅由权利要求和它们的等同物限定。
实施例1
本实施例提供一种16α,16β,17β-三氘-1,4-雄烯二酮的合成方法,所述合成方法包括以下步骤:
(a1)1,4-雄烯二酮与氘代水在四氢呋喃中混合,在催化量的弱碱性盐(如碳酸钾、碳酸钠)的催化作用下加热回流24-30小时;所述1,4-雄烯二酮与氘代水的物质的量比为1:100~1:500(如1:300),所述四氢呋喃的体积为氘代水体积的1/2~1/4(如1/3);
(a2)停止回流,冷却至室温,采用乙酸乙酯萃取回流反应液,对萃取后的有机相进行浓缩处理获得中间产物16α,16β–二氘-1,4-雄烯二酮;
(a3)所述中间产物与硼氘化钠在氘代甲醇中混合,在-10℃~0℃的冰浴条件下反应20-60分钟;所述中间产物与硼氘化钠的物质的量比为1:1~1:1.2(如1:1.1);
(a4)用盐酸或硫酸酸化还原反应液使其ph≤2,避免还原后17位上的-oh变成-ona;
(a5)采用乙酸乙酯萃取酸化后的还原反应液,并用柱层析法分离有机相,获得目标产物。
图1给出了目标产物的1hnmr图谱,从图中可以看出明显的3个双键氢,并且氢的数量正好跟16α,16β,17β-三氘-1,4-雄烯二酮的氢数量完全对应;图2给出了目标产物的13cnmr图谱,从图中可以看到明显的一个羰基碳和四个双键碳,也能够看到两个连有氘原子的碳的裂分,并且碳的数量正好跟16α,16β,17β-三氘-1,4-雄烯二酮的碳数量完全对应。因此,可以确认目标产物即为16α,16β,17β-三氘-1,4-雄烯二酮。
本实施例的优势在于:合成路线简单,反应条件温和,原料廉价易得、成本低。
实施例2
本发明实施例1的具体应用例。
(1)称取1.42g1,4-雄烯二酮放入到三口瓶中,加入催化量碳酸钾50mg,量取30ml纯度为99.9%氘代水和10ml四氢呋喃共同加入到反应液中,加热回流24小时后停止反应,冷却至室温;
(2)反应液用40ml乙酸乙酯萃取三次,有机相收集到一起用旋转蒸发仪旋蒸,得到白色固体粉末1.28g,反应收率为90%;
(3)将所述1.28g白色固体粉末加入三口瓶中,称取硼氘化钠207mg放入三口瓶,加入18ml氘代甲醇作为反应溶剂,将所述三口瓶置于冰浴条件下(0℃)反应30分钟;
(4)停止反应,向还原反应液中滴加1mol/l的盐酸直至ph≤2;
(5)用20ml乙酸乙酯萃取三次,留下有机相,用正己烷:乙酸乙酯=1:1展开剂进行过柱分离,用旋转蒸发仪去除有机物后获得白色产物0.75g,收率为59%。
技术特征:
技术总结
本发明涉及一种16α,16β,17β‑三氘‑1,4‑雄烯二酮的合成方法,所述合成方法包括以下步骤:(A1)1,4‑雄烯二酮与氘代水在第一溶剂中混合,并在弱碱性盐的催化作用下加热回流;(A2)萃取回流反应液,浓缩萃取的有机相后得到中间产物16α,16β–二氘‑1,4‑雄烯二酮;(A3)所述中间产物与硼氘化钠在第二溶剂中混合,并在冰浴条件下进行选择性还原;(A4)酸化还原反应液使其pH≤2;(A5)萃取酸化后的还原反应液,采用柱层析法分离有机相,获得目标产物16α,16β,17β‑三氘‑1,4‑雄烯二酮。本发明具有反应路线简单、产物纯度高、成本低等优点。
技术研发人员:赵鹏;陈武炼;张驰中
受保护的技术使用者:上海安谱实验科技股份有限公司
技术研发日:2017.03.17
技术公布日:2017.07.14
- 还没有人留言评论。精彩留言会获得点赞!