合成5,6,4'‑三羟基黄酮‑7‑0‑D‑葡萄糖醛酸有关物质及其制备方法和应用与流程
本发明涉及药物化学技术领域,尤其涉及一种合成5,6,4'-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质及其制备方法和应用。
背景技术:
5,6,4'-三羟基黄酮-7-0-d-葡萄糖醛酸,又名灯盏花乙素、野黄芩苷,是从菊科飞蓬属植物短亭飞蓬(eriger0nbreviscapus)的全草(又名灯盏细辛)中提取的黄酮类混合物的主要有效成分,分子式为c21h18012,分子量为462.37,其结构式如式ⅳ所示:
临床研究表明5,6,4'-三羟基黄酮-7-0-d-葡萄糖醛酸对高血压、脑出血、脑血栓形成,脑栓塞多发性神经炎,慢性蛛网膜炎及其后遗症具有较好疗效,并对风湿病、冠心病也有一定疗效。此外大量的研究证实5,6,4'-三羟基黄酮-7-0-d-葡萄糖醛酸具有抗炎、抗氧化、蛋白激酶抑制剂作用,还具有对糖尿病、肾病、风湿类风湿等疾病的治疗作用。因此,目前5,6,4'-三羟基黄酮-7-0-d-葡萄糖醛酸临床上广泛用于治疗脑血栓、脑梗塞、多发性神经炎、慢性蛛网膜等脑血管意外所致瘫痪以及冠心病、心绞痛、治疗痛风性关节炎等症。
目前除了植物提取的合成5,6,4'-三羟基黄酮-7-0-d-葡萄糖醛酸,合成5,6,4'-三羟基黄酮-7-0-d-葡萄糖醛酸作为改良型创新原料药,具有良好的市场前景。但是合成5,6,4'-三羟基黄酮-7-0-d-葡萄糖醛酸杂质谱不同于目前上市的植物提取5,6,4'-三羟基黄酮-7-0-d-葡萄糖醛酸,对于合成5,6,4'-三羟基黄酮-7-0-d-葡萄糖醛酸的有关物质目前尚无研究。杂质的存在将影响产品使用的安全性,按照新药开发过程中杂质控制的相关指导原则的要求,应对样品中杂质进行彻底研究,严格控制。
技术实现要素:
有鉴于此,本发明要解决的技术问题在于提供一种合成5,6,4'-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质及其制备方法和应用。
本发明提供的合成5,6,4'-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质或其盐,结构如式(ⅰ)所示:
本发明所述的合成5,6,4'-三羟基黄酮-7-0-d-葡萄糖醛酸是指通过化学合成的方法制备的5,6,4'-三羟基黄酮-7-0-d-葡萄糖醛酸。具体制备方法参照昆药集团申请号分别为cn201210114758.9,cn201210114894.8,pct/cn2012/086543的专利申请。
所述合成5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸含量大于99%,其中含有微量的合成5,6,4'-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质,含量低于0.5%。
本发明还提供了上述合成5,6,4'-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质的制备方法,包括以下步骤:
1)式(ⅱ)所示化合物在催化剂的作用下,进行选择性消除反应,生成式(ⅲ)所示化合物;所述催化剂为1,8-二氮杂二环十一碳-7-烯(dbu)、吡啶和4-二甲氨基吡啶(dmap)中的任意一种或几种;
2)式(ⅲ)所示化合物进行水解反应,得到式(ⅰ)所示化合物;
其反应方程式如下:
本发明首先以1,8-二氮杂二环十一碳-7-烯(dbu)、吡啶和4-二甲氨基吡啶(dmap)中的任意一种或几种作为催化剂,式(ⅱ)所示化合物在催化剂的作用下,进行选择性消除反应,选择性的脱去一分子水,生成式(ⅲ)所示化合物。上述催化剂有助于提高选择性消除反应的速率和选择性。
所述选择性消除反应优选在溶剂中进行,在本发明的某些具体实施例中,所述溶剂选自二氯甲烷、三氯甲烷、乙腈、甲苯、四氢呋喃和1,4-二氧六环中的任意一种或几种。
在本发明的某些具体实施例中,所述式(ⅱ)所示化合物与催化剂的摩尔比为1:(0.1~5),在某些实施例中为1:(0.2~3),在一些实施例中为1:1。
所述选择性消除反应的温度为10~30℃,反应时间为3~20h,在某些实施例中为4~6h,在一些实施例中为8h,在一些实施例中为10h。
优选的,所述步骤2)具体为:式(ⅲ)所示化合物在无机碱催化下进行水解反应,然后加入无机酸调节体系ph值至2~3,进行中和反应,得到式(ⅰ)所示化合物。
在本发明的某些具体实施例中,所述无机碱选自氢氧化钠、氢氧化锂、氢氧化钾、碳酸钾和碳酸钠中的任意一种或几种。
所述无机酸选自盐酸、硫酸、乙酸、甲酸、硝酸和磷酸中的任意一种或几种。
所述水解反应的溶剂选自丙酮、水、甲醇、乙醇、二氯甲烷、二甲基亚砜、四氢呋喃和二氧六环中的任意一种或几种。
所述水解反应的温度优选为-10~40℃,在某些实施例中为-8~0℃,在一些实施例中为-5℃;反应时间优选为1~10h,在某些实施例中为2~4h,在一些实施例中为3h,在一些实施例中为2h,在一些实施例中为4h。
所述式(ⅲ)所示化合物与无机碱的摩尔比优选为1:(0.1~50),在某些实施例中为1:(10~30),一些实施例中为1:25,一些实施例中1:15,一些实施例中1:20。
所述中和反应的温度优选为-10℃~40℃,在一些实施例中为-5℃。
在本发明的某些具体实施例中,可以进一步采用常规方法如重结晶进行提纯,得到符合要求纯度的杂质对照品。
本发明通过hplc法、uv吸收法、核磁共振、ms谱分析对上述合成5,6,4'-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质进行了结构验证。
本发明所述5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质的结构确证包括以下步骤:
步骤1、hplc法证实制备所得5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质b与昆药集团股份有限公司合成5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸中有关物质(杂质)b的保留时间完全一致。
步骤2、uv谱图证实制备所得5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质b与昆药集团股份有限公司合成5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸中有关物质b特征峰完全一致。
步骤3、核磁共振氢谱、碳谱对制备所得5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质b的化学结构进行了确证。
步骤4、经ms谱分析,证实昆药集团股份有限公司合成5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸中有关物质b,与制备所得5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质b,具有相同的分子离子信号。
本发明所述结构确证方法步骤1中,色谱条件流动相和体积比为:流动相a:water/tfa=1000/1(v/v),流动相b:acet0nitrile/tfa=1000/1(v/v),样品溶剂:acetonitrile/dmso=50/50(v/v)。
本发明所述结构确证方法步骤3和步骤4证实了5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸杂质b的化学结构如式ⅰ所示。
本发明还提供了上述合成5,6,4'-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质或其盐或上述制备方法制备的合成5,6,4'-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质,在合成5,6,4'-三羟基黄酮-7-0-d-葡萄糖醛酸或其制剂有关物质检测时,作为对照品的应用。
具体的,可应用于合成5,6,4'-三羟基黄酮-7-0-d-葡萄糖醛酸或其制剂的杂质分析控制、药理毒理研究等。
本发明以5,6,4’-三乙酰氧基黄酮-7-o-d-三乙酰氧基葡萄糖醛酸甲酯(结构式如式ⅱ所示)为原料,经选择性消除和水解两步反应,成功制备得到5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质b,并进一步纯化,最终获得5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质b单体,进行确证后得出该有关物质化学结构。该方法合成路线短、成本低、操作简单,结果准确,产品纯度高,反应收率高,产品易于纯化,可用于合成5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸样品的杂质分析控制,有利于合成5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸的质量控制,更好的保障了产品质量。
附图说明
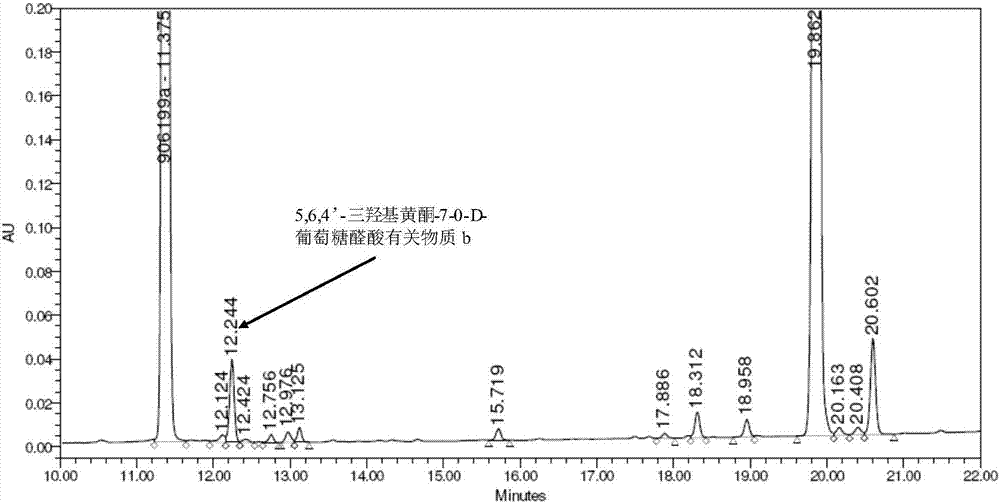
图1是实施例1制备的5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质b(批号:20140913)的hplc分析谱图;
图2是昆药集团股份有限公司合成的5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸(批号20140424-1)的hplc分析谱图;
图3是实施例1制备的5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质b(批号20140913)以5%质量分数加入到5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸(批号20140424-1)的hplc分析谱图;
图4是昆药集团股份有限公司合成的5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸(批号20140424-1)中5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质b的uv谱图;
图5是实施例1制备的5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质b(批号20140913)的uv谱图;
图6是实施例1制备的5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质b(批号20140913)的hplc分析谱图;
图7是合成5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸(批号20140424-1)+实施例1制备的5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质b(20140913)的ms谱图;
图8是实施例1制备的5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸杂质b(20140913)的ms谱图;
图9是合成5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸(批号20140424-1)的ms谱图。
具体实施方式
为了进一步说明本发明,下面结合实施例对本发明提供的合成5,6,4'-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质(以下简称5,6,4'-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质b)及其制备方法和应用进行详细描述。
实施例1
(1)化合物ⅲ的制备
20g(27.4mmol)5,6,4’-三乙酰氧基黄酮-7-o-d–三乙酰氧基葡萄糖醛酸甲酯溶解于200ml甲苯中,然后搅拌下缓慢滴加入1,8-二氮杂二环十一碳-7-烯(dbu)4.1g(27mmol),30℃下搅拌反应约8小时反应完全。反应混合物搅拌下加入约500ml乙酸乙酯,用500ml水洗两次。分出水层后的有机层中加入适量无水硫酸钠干燥,减压蒸干。硅胶柱层析并用石油醚:乙酸乙酯=5:1(v/v)的洗脱剂洗脱,得15g白色固体化合物ⅲ,色谱纯度98.00%以上,收率为82%。
1h-nmr(400mhz,dms0),δ(ppm):7.95(2h,d,j=8.6hz),7.51(1h,s),7.32(2h,d,j=8.6hz),6.78(1h,s),6.15(1h,d,j=7.8hz),5.57(1h,d,j=10hz),5.38(1h,m),4.94(1h,m),3.67(3h,s),2.34(3h,s),2.31(3h,s),2.25(3h,s),2.02(3h,s),2.01(3h,s).
(2)5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质b的制备
13.4g(20mmol)化合物ⅲ中加入200ml丙酮和200ml水的混合溶剂,在-5℃搅拌下缓慢滴加10%的naoh水溶液200ml,搅拌反应约三小时后,经tlc监测反应已完全。-5℃下缓缓向反应液中滴加2mol/l盐酸水溶液中和反应液至ph为2~3,持续搅拌一小时并逐渐升至室温,反应液中渐渐有黄色固体5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸析出。室温下静置过夜,过滤,滤饼用适量水和丙酮洗,干燥后用乙醇重结晶得到8g5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质b,批号记为20140913,收率为89%。hplc检测,纯度为98.88%,hplc检测的色谱图和统计结果见图1和表1,表1是图1的数据汇总。
1h-nmr(400mhz,dms0),δ(ppm):12.78(1h,s),10.51(1h,s),8.71(1h,s),7.93(2h,d,j=8.8hz),7.11(1h,s),6.94(2h,d,j=8.8hz),6.83(1h,s),5.99(1h,d,j=3.6hz),5.81(1h,d,j=5.6hz),5.74(1h,s),5.28(1h,s),4.14(1h,dd,j=4.0,4.5hz),3.84(1h,dd,j=4.5,6.0hz).
表1图1的数据汇总
实施例2
(1)化合物ⅲ的制备
20g(27.4mmol)5,6,4’-三乙酰氧基黄酮-7-o-d–三乙酰氧基葡萄糖醛酸甲酯溶解于200ml二氯甲烷中,然后搅拌下缓慢加入4-二甲氨基吡啶(dmap)1.7g(13.7mmol,0.5eq),25℃下搅拌反应约10小时反应完全。反应混合物搅拌下加入约300ml乙酸乙酯,用500ml水洗两次。分出水层后的有机层中加入适量无水硫酸钠干燥,减压蒸干。硅胶柱层析并用石油醚:乙酸乙酯=5:1(v/v)的洗脱剂洗脱,得14.3g白色固体化合物ⅲ,色谱纯度98.00%以上,收率为78%。核磁鉴定结果与实施例1结果一致。
(2)5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质b的制备
13.4g(20mmol)化合物ⅲ中加入200ml丙酮和200ml水的混合溶剂,在0℃搅拌下缓慢滴加10%的氢氧化钾水溶液220ml,搅拌反应约四小时后,经tlc监测反应已完全。0℃下缓缓向反应液中滴加乙酸中和反应液至ph为2~3,持续搅拌一小时并逐渐升至室温,反应液中渐渐有黄色固体5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质b析出。室温下静置过夜,过滤,滤饼用适量水和丙酮洗,干燥后用乙醇重结晶得到7.4g5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质b,收率为83%。hplc检测,纯度达98.00%以上,核磁鉴定结果与实施例1结果一致。
实施例3
(1)化合物ⅲ的制备
20g(27.4mmol)5,6,4’-三乙酰氧基黄酮-7-o-d–三乙酰氧基葡萄糖醛酸甲酯溶解于200ml四氢呋喃中,然后搅拌下滴加入吡啶6.5g(82.2mmol),25℃下搅拌反应约6小时反应完全。反应混合物搅拌下加入约200ml乙酸乙酯,用500水洗两次。分出水层后的有机层中加入适量无水硫酸钠干燥,减压蒸干。硅胶柱层析并用石油醚:乙酸乙酯=5:1(v/v)的洗脱剂洗脱,得14.7g白色固体化合物ⅲ,色谱纯度98.00%以上,收率为80%。核磁鉴定结果与实施例1结果一致。
(2)5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质b的制备
13.4g(20mmol)化合物ⅲ中加入200ml丙酮和200ml水的混合溶剂,在5℃搅拌下缓慢滴加10%的氢氧化锂水溶液190ml,搅拌反应约两小时后,经tlc监测反应已完全。5℃下缓缓向反应液中滴加2mol/l硫酸水溶液中和反应液至ph为2~3,持续搅拌一小时并逐渐升至室温,反应液中渐渐有黄色固体5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质b析出。室温下静置过夜,过滤,滤饼用适量水和丙酮洗,干燥后用乙醇重结晶得到7.6g5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质b,收率为85%。hplc检测,纯度达98.00%以上,核磁鉴定结果与实施例1结果一致。
实施例4结构确证
1、hplc确证合成制备所得化合物为合成5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸中的5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质b:
hplc色谱条件如下:
hplc系统:watersalliance2695四元泵
watersalliance2998pda检测器
watersalliance2695自动进样器
watersalliance2695柱温箱
empower2网络版工作站
色谱柱:agilentzorbaxsb-c8,5um,250x4.6mmpn:880975-906
流速:1.0ml/min.
检测波长:uv254nm
进样体积:10ul
柱温:30℃
自动进样器温度:5℃
流动相a:water/tfa=1000/1(v/v)
流动相b:acetonitrile/tfa=1000/1(v/v)
样品溶剂:acetonitrile/dms0=50/50
样品浓度:1.0mg/ml
实施例1合成制备获得的5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质b(批号:20140913),与合成5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸(批号20140424-1)中含有的杂质b的等同性验证:
分别对合成制备所得5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质b、合成5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸进行hplc分析(谱图如图1、图2所示),表2是图2的数据汇总。再将约5%的5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质b标准品,加入昆药集团股份有限公司合成的5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸样品(批号20140424-1),进行hplc分析(谱图如图3所示),表3是图3的数据汇总。
分析结果显示,合成制备获得的5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质b标准品出峰位置与5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸中本身含有的5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质b完全吻合。
对二者的uv吸收进行检测,谱图如图4、图5所示,结果表明两者的uv吸收图谱完全一致。因此它们的等同性可以得到确认。
表2图2的数据汇总
表3图3的数据汇总
2、质谱和lc/ms解析
测试条件:
质谱:waterssqd
软件:watersempower2网络版
source:120℃desolvati0n:300℃capilary:3.5kv
lc-ms:
流动相a:超纯水(milliporeultrapurewatersystem,resistivity:18.2mω.cm)+0.1%甲酸
pumpb:色谱纯乙腈(merckkgaa64271,hypergradeforlc-ms)+0.1%甲酸
色谱柱:
柱温:30℃
检测波长:λ=254nm,230nmand218nm/pda
流速:1.0ml/min
谱图见图6、图7、图8和图9。
由上述谱图可知,对5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸(批号20140424-1)中含有的杂质b,以及合成制备的5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸杂质b(批号201409132)进行lc/ms分析,均显示其分子量为444([m-h]-443)(见图8和图9)。由其分子量可以推测,杂质b应该是5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸脱一分子水后的产物。并且,5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸中本身含有的杂质b,与合成制备的杂质b,具有相同的分子离子信号。
3、5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸杂质b的核磁共振谱图解析
核磁共振氢谱:
仪器型号:brukeravanceiii400spectrometer(z115310,sn10032129)
溶剂:[d6]dmso(d,99.9%)+0.03%v/vtms,10*0.6mlampoule,cambridgeisotopelaboratories,inc.,lot12a-029
温度:25℃
测试数据见表4。
表4核磁共振氢谱数据汇总
核磁共振13c谱:
仪器型号:brukeravanceiii400spectrometer(z115310,sn10032129)
溶剂:[d6]dmso(d,99.9%)+0.03%v/vtms,10*0.6mlampoule,cambridgeisotopelaboratories,inc.,lot12a-029
温度:25℃
结果见表5。
表55,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸杂质b的核磁共振碳谱分析
由以上结果可知,ms一级谱给出了杂质准确的分子量信息:m/z442.7,与5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸脱一分子水后所得的5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质b分子量一致。hplc谱确证了制备所得5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质b与合成5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸中5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质b的保留时间完全一致,且uv谱证实了二者的特征吸收峰完全一致。核磁共振氢谱和碳谱,证实了5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质(杂质)b化学结构如式(ⅰ)所示:
由上述实施例可知,本发明证实了上述5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸有关物质b的结构,并成功制备得到该化合物,且具有较高的纯度,可以直接用于合成5,6,4’-三羟基黄酮-7-0-d-葡萄糖醛酸的杂质分析控制,以此基础制订了严格的杂质限度,为将来产品的安全使用提供了理论依据。
以上实施例的说明只是用于帮助理解本发明的方法及其核心思想。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!
- 包含甲氧基黄酮的NOX抑制剤及NFκB抑制剤的制作方法与工艺
- 一种多甲氧基黄酮、组合物及其制剂用于预防或治疗糖尿病的用途的制作方法与工艺
- 一种1‑(2‑羟基‑3‑甲氧苯基)乙酮的制备工艺的制作方法与工艺
- 一种多甲氧基黄酮在制备预防心血管炎症药物上的应用的制作方法与工艺
- 3’-羟基染料木黄酮用于制备抑制黑色素生成的组合物的用途的制作方法与工艺
- 合成5,6,4'‑三羟基黄酮‑7‑0‑D‑葡萄糖醛酸有关物质及其制备方法和应用与流程
- 5‑(4‑羟基‑3‑甲氧基苯亚甲基)‑2‑(2‑硝基苯亚胺基)噻唑烷酮及应用的制作方法与工艺
- 制备荷羟酮(甲基)丙烯酸酯的方法与流程
- 从瓯柑果实中分离纯化多甲氧基黄酮类化合物的方法与流程
- 一种多甲氧基黄酮在制备治疗黑色素瘤的药物上的应用的制造方法与工艺