含金属晶体海藻酸单体的制造方法与流程
本发明涉及一种海藻酸单体结构的制造方法,且特别是一种含金属晶体海藻酸单体结构的制造方法。
背景技术
海藻酸(alginate)为一种水溶性天然多醣体高分子,其主要原料是由海洋中褐藻类所萃取的海藻酸钠。海藻酸的单体结构主要是由古罗醣醛酸(α-l-guluronate,分子式为c6h8o6,代号为:g)与其立体异构物甘露醣醛酸(β-d-mannuronate,分子式为c6h8o6,代号为:m)两种醣醛酸分子以m-m、g-g、或m-g的组合方式经由1,4糖苷键相连形成一种无支链的线性嵌段共聚物,海藻酸的分子量范围约在1万到60万。由于海藻酸为阴离子型高分子,所以海藻酸容易与带正电荷的金属离子产生交联反应而形成非水溶性的海藻酸凝胶,其中特别是带有二价正电荷的阳离子,例如钙离子(ca2+)、钡离子(ba2+)、锌离子(zn2+)等。海藻酸凝胶具有热不可逆性,并且可藉由改变海藻酸凝胶中g/m的含量比值来调整其柔韧度与硬度。由于海藻酸凝胶为天然多醣体高分子,其经过灭菌或抗菌处理后可广泛地被应用于食品、纺织以及生医等产业。
例如在纺织以及生医等产业中,应用海藻酸凝胶所制成的海藻酸纤维具有良好的生物相容性以及螯合重金属离子的能力,如海藻酸纤维中更添加各种营养或抗菌成分,则由其所制成的功能性织物更可具有保养、美容或医疗等效能。举例而言,银离子具有很强的抗菌力,含银离子的海藻酸纤维所制成的医疗用敷布,对于伤口除具有良好的抗敏性、防护性,更有助于伤口的复原。目前制造含银离子的海藻酸纤维主要有涂布含银离子化合物于海藻酸纤维上;或于制造海藻酸纤维的过程中添加含银离子化合物等方法。然而,上述各种方法均系藉由银离子随机结合于海藻酸纤维表面结构中,且银离子容易因氧化而失去抗菌力。此外,上述方法须于海藻酸纤维中涂布高浓度的银离子方得达到医疗敷布应有的效果,但同时也会有较高生物毒性的副作用及无法使用于人体的风险。
目前业界已经有提出先制作含银海藻酸单体的想法,使之后制作的含银海藻酸纤维能具有银离子均匀分布的特性,以改善上述问题。但由于海藻酸天糖体高分子的特性,难以控制银离子与海藻酸单体的结合方式以及银离子的分布,导致了制程上的困难。因此,有必要针对上述问题提出一种含金属晶体的海藻酸单体的制造方法。
技术实现要素:
本发明提供一种含金属晶体海藻酸单体结构的制造方法,包括以下步骤:(s1)提供海藻酸高分子溶液,包括具有数个海藻酸单体;(s2)加入自动还原剂与金属前驱物于海藻酸高分子溶液中,形成混合溶液,自动还原剂占混合溶液0.05%~0.2%(w/v,或以g/l表示),且金属前驱物占混合溶液的0.1~6mm;以及(s3)微波加热混合溶液5~30分钟。
在本发明的部分实施例中,其中金属前驱物具有的金属离子为以下任一者或其组合:钙离子、钡离子、铜离子、银离子、金离子、铁离子以及锌离子。
在本发明的部分实施例中,其中金属驱物为以下任一者或其组合:氯金酸、硝酸银、硝酸锌、碳酸银、醋酸锌、硫酸铜、硫酸铁、硫酸锌。
在本发明的部分实施例中,步骤(s2)之后、步骤(s3)之前还包括步骤:(s2-1)将混合溶液混合1分钟~10分钟。
在本发明的部分实施例中,其中步骤(s2-1)是以转速700~900rpm进行混合。
在本发明的部分实施例中,其中步骤(s3)的微波加热功率为700w~900w。
在本发明的部分实施例中,其中步骤(s3)的微波加热功率为900w且时间介于5~15分钟。
在本发明的部分实施例中,其中步骤(s2)包括以下次步骤:(s21)加入金属前驱物于海藻酸高分子溶液中,以转速700rpm混合5~15分钟;以及(s22)加入自动还原剂于海藻酸高分子溶液中,形成混合溶液。
在本发明的部分实施例中,其中步骤(s2)与步骤(s3)之间隔时间小于等于1小时。
在本发明的部分实施例中,其中步骤(s3)中,会形成数个金属晶体于数个海藻酸单体中,且金属晶体的粒径介于20nm~712nm之间。
在本发明的部分实施例中,其中步骤(s3)中,会形成数个金属晶体于数个海藻酸单体中,且金属晶体的粒径介于20nm~255nm之间。
在本发明的部分实施例中,其中海藻酸单体具有以下任一或多种结构:古罗醣醛酸-甘露醣醛酸(g-m)、甘露醣醛酸-甘露醣醛酸(m-m)、古罗醣醛酸-古罗醣醛酸(g-g)。
在本发明的部分实施例中,其中步骤(s2)形成的混合溶液具有ph值介于6~7之间。
在本发明的部分实施例中,其中自动还原剂具有ph值介于4~6之间。
在本发明的部分实施例中,其中自动还原剂选自过氧化氢、硼氢化钠、抗坏血酸、葡萄糖(glucose)、淀粉、羧甲基纤维素、柠檬酸、亚硫酸盐(sulphites)、多酚类(polyphenols)或上述任意组合。
在本发明的部分实施例中,其中自动还原剂为过氧化氢。
在本发明的部分实施例中,本发明提供的制造方法具有一总制程时间小于等于1.5小时。
在本发明的部分实施例中,其中步骤(s2)中还包括加入金属化合物,具有与该金属前驱物相同的金属源。
本发明因修饰海藻酸单体的分子结构形成羰基,故可使金属晶体稳定地形成于海藻酸单体以及/或是海藻酸盐单体之中,而形成含金属晶体海藻酸单体结构以及含金属晶体海藻酸盐单体结构。相较于现有技术,本发明提供的方法所制作出来的含金属海藻酸单体可以良好控制金属晶体于海藻酸单体上的结合方式,并且能依此提供具有较低金属晶体浓度且能长时间稳定释出金属晶体/离子的海藻酸纤维,达到医疗敷料的效果,同时又能避免现有技术的生物毒性的副作用,所以能广泛地应用于食品、纺织以及生医等产业。
附图说明
为让本发明的上述和其他目的、特征和优点能更明显易懂,下文特举数个较佳实施例,并配合所附图式,作详细说明如下。
图1为具有g-g结合方式的海藻酸单体结构的示意图。
图2为依据本发明提供的方法制作出的具有g-g结合方式的含金属海藻酸单体结构的示意图。
图3为依据本发明提供的方法制作出的具有g-m结合方式的含金属海藻酸单体结构的示意图。
图4为依据本发明提供的方法制作出的具有m-m结合方式的含金属海藻酸单体结构的示意图。
具体实施方式
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
本发明提供一种含金属晶体海藻酸单体结构的制造方法,能提供高稳定度的含金属晶体海藻酸单体结构,其具有的金属晶体能稳定地键结于海藻酸单体上,且使用本发明提供的海藻酸单体制作出的海藻酸纤维能具有均匀的金属晶体的特性。相较于现有技术,本发明提供的方法所制作出来的含金属海藻酸单体可以良好控制金属晶体于海藻酸单体上的结合方式,并且能依此提供具有较低金属晶体浓度且能稳定释出金属晶体/离子(部分以晶体形式释出,部分以离子形式释出),的海藻酸纤维,达到医疗敷料的效果,同时又能避免现有技术的生物毒性的副作用。为让本发明的上述和其他目的、特征和优点能更明显易懂,下文以实施例做详细说明,以使本发明的组合物、应用方法与功效能更容易理解,但并非用以限制本发明。
本发明中下文所述的海藻酸单体由两个醣醛酸分子组合而成,两个醣醛酸分子可分别选自古罗醣醛酸(α-l-guluronate;代号为:g)或甘露醣醛酸(β-d-mannuronate;代号为:m),组合方式可以是m-m、g-g、或m-g。
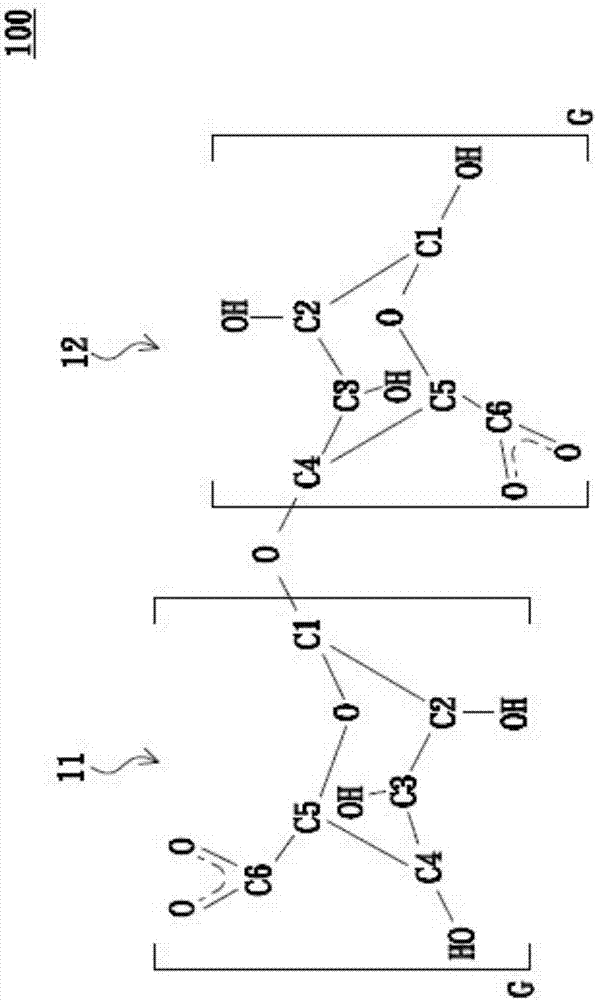
依据本发明提供的制造方法,首先提供海藻酸高分子溶液,其所包括的海藻酸高分子可选用萃取天然褐藻中海藻酸钠(sodiumalginate)成份制备而得的市售品。海藻酸高分子由数个海藻酸单体经由数个分子间糖苷基聚合而成,海藻酸单体可以是g-g、g-m及/或m-m混合的结合方式。海藻酸高分子占海藻酸高分子溶液的0.25~0.5wt%(体积百分比),且依据不同实施例可以选择以g-g、g-m或/及m-m混合的结合方式,并且m/g的比例没有限制。以下为方便说明,仅以g-g结合方式的海藻酸单体举例做说明之用,但并非用以限制本发明,于本发明其他实施例中,可以使用g-g、g-m、m-m或上述任意组合的海藻酸高分子溶液。如图1所示为g-g结合方式的海藻酸单体100,醣醛酸分子11与醣醛酸分子12共同形成海藻酸单体100,分子结构中的碳以c1~c6表示,其中醣醛酸分子11与12上的c2与c3均为羟基(c-oh)。
将自动还原剂与金属前驱物分别加入海藻酸高分子溶液中(加入方式不限制,可以直接倒入或是滴定等方式),形成混合溶液。混合溶液的ph值介于6~7之间;自动还原剂较佳为弱酸性(ph值介于4~6之间),可以为过氧化氢(hydrogenperoxide)、硼氢化钠(nabh4;sodiumborohydride)、抗坏血酸(ascorbicacid)、葡萄糖(glucose)、淀粉(starch)、羧甲基纤维素(cmc)、柠檬酸(citrate)、亚硫酸盐(sulphites)、多酚类(polyphenols)或上述任意组合,其中以过氧化氢为佳,并且本案的自动还原剂占混合溶液0.05%~0.2%(重量体积比w/v,或是以g/l表示);金属前驱物占混合溶液的0.1~6mm(毫摩尔体积浓度),并且金属前驱物所含的金属较佳选自过渡金属元素,如:钙(ca)、钡(ba)、铜(cu)、银(ag)、金(au)、铁(fe)、锌(zn),由于不同金属具有不同程度的抗菌功效,例如铜可以应用于子宫内投药避孕,因此金属前驱物的所包括的金属离子选择可依据不同应用做选择;金属前驱物较佳选自氯金酸、硝酸银、硝酸锌、碳酸银、醋酸锌、硫酸铜、硫酸铁、硫酸锌、其他包括上述金属元素的化合物中的任一者或其组合。由于海藻酸高分子溶液中的海藻酸高分子通常黏稠,因此选择性将混合溶液以700~900rpm之间的转速混合1分钟~10分钟,以使混合溶液中的自动还原剂与金属前驱物均匀混合。之后将混合溶液以功率700w~900w微波加热5~30分钟,较佳功率900w且时间介于5~15分钟,得到含有含金属晶体海藻酸单体结构的成品溶液,如图2所示的海藻酸单体结构101,其包括的醣醛酸分子11与醣醛酸分子12的c2上的羟基分别被氧化成羰基(c=o)112与121,并且c6上的羧基122(羧基上两个氧原子间的虚线符号表示自由价电子于两个氧原子间的共振态)能协同稳定金属晶体13(值得注意的是,图2同样以g-g结合方式为例绘示,仅做说明之用,并非用以限制本发明;另外,一般来说仅会有单一金属晶体13形成醣醛酸分子11与醣醛酸分子12之间,但金属晶体13并非限定键结于醣醛酸分子11的羰基112,于其他实施例中,金属晶体13可以配位键结于羰基121,羧基112能协同稳定金属晶体13)。
自动还原剂与金属前驱物的加入顺序不限,可以先加入自动还原剂、先加入金属前驱物,或是两者同时加入。但由于自动还原剂有可能会对海藻酸单体100造成破坏,因此于加入自动还原剂之后到微波加热的间隔时间较佳不超过1小时。
以先加入金属前驱物的实施样态为例说明,依据本发明的一实施例将金属前驱物加入海藻酸高分子溶液中,以转速700rpm混合5~15分钟,之后加入自动还原剂后直接进行微波加热。在反应过程中,自动还原剂会对海藻酸单体进行水解和氧化,以图1所示的g-g海藻酸单体100为例说明,醣醛酸分子11与12上的部份c2与c3上的会羟基(c-oh)氧化形成羰基(c=o)。而依据本案发明人实际研究后发现,c2上的羟基氧化形成羰基的比例会远高于c3上的羟基氧化形成羰基,因此图2中仅绘示出醣醛酸分子11的c2上的羟基氧化形成羰基111,以及醣醛酸分子12的c2上的羟基氧化形成羰基121。金属前驱物于混合溶液中解离,形成带正电的金属离子游离于混合溶液中,金属离子受到带负电的羰基111,形成配位键结于羰基111的氧上。此时于混合溶液中,有部分金属离子配位结合于海藻酸单体上,并且有部分金属离子以离子形态游离于混合溶液中。接着,混合溶液中部分游离的金属离子作为材料来源,在羧基122的稳定协同作用下,自动还原剂还原羰基111上键结的金属离子,使其成为键结型金属晶种,同时还原部分游离的金属离子共聚于金属晶种上,形成金属晶体13固定于海藻酸单体结构101中,如图2所示。而溶液中其他未形成金属晶体13的游离金属离子则会被还原成金属原子。
上述说明中有提及,由于c2上的羟基氧化形成羰基的比例会远高于c3上的羟基氧化形成羰基,因此图式仅绘示出c2具有羰基作为说明。而在c3上的羟基氧化形成羰基的状况下(未绘示出),c3羰基亦能协同稳定金属晶体13于羰基111上,因此对于含金属海藻单体结构101具有更稳定的效果。
图3所示为依据本发明上述相同方法制作出的含金属海藻酸单体结构102,其中海藻酸单体结构具有g-m结合方式;而图4所示为依据上述相同方法制作出的含金属海藻酸单体结构103,其中海藻酸单体结构具有m-m结合方式。为方便说明与容易理解,图3~图4具有相同功能的基团和分子,沿用与图1~图2相同的元件标号。
而于金属前驱物与自动还原剂同步加入的实施样态中,以及先加入自动还原剂后加入金属前驱物的实施样态中,实验条件同上说明,可以得到如图2所示的含金属海藻单体结构。
再者,依据本发明不同实施例,于微波加热前还可以选择性加入包括与金属前驱物相同金属源的金属化合物作为辅助剂,金属化合物可与自动还原剂形成共轭氧化还原剂,其中所加入的金属化合物占混合溶液的50~250ppm。
值得注意的是,依据本发明提供的方法,最终产物(包括于成品溶液中的成分)不限于上述的含金属海藻酸单体结构,尚可包括甘油酸(glycerate)或丙酮酸(pyruvate)生成,其来源系由于使用甘露醣醛酸的实施例中,部份甘露醣醛酸经水解和氧化所致,其水解程度视所加入的自动氧化剂比例而成正向关系,在此不做限制。
由上述说明可知,依据本发明的方法,于海藻酸高分子中所形成金属晶体可稳定地结合于经设计修饰后的海藻酸单体结构中。而使用本发明制作出的含金属海藻酸单体结构来制作出的含金属海藻酸纤维,亦能具有稳定且良好的抗菌效果。再者,本发明方法制作出的含金属海藻酸单体结构,其包括的金属晶体具有20nm~712nm之间的粒径,且较佳介于20nm~255nm之间。金属经体粒径可以经由动态光散射粒径分析仪及界面电位分析仪进行量测,并且可以藉由调整成份比例、微波功率以及微波时间,有效控制金属晶体尺寸,使金属晶体尺寸落在适用于人体且具有良好抗菌效果的范围内(金属晶体尺寸过小会有生物毒性,进入人体细胞;但尺寸过大又会无法通过细菌细胞壁,导致无法达到良好的抗菌效果),进而更进一步控制金属晶体于依据其所制作出的敷料中的浓度。本发明提供的方法能制作出稳定的含金属海藻酸单体结构,配合控制金属晶体尺寸,能相较于现有技术降低生物毒性,能适用于人体。并且,依据本发明上述方法,能控制混合溶液温度在40℃~60℃之间即可完成含金属海藻酸单体结构的制程,并且总制程时间可以控制在不超过1.5小时,一般来说可以控制在1小时内,较佳可以控制在15分钟内,且更佳可以控制在10分钟内,大幅提高制程效率,减少制程与人力成本。
为使本发明的功效更容易理解,以下提供具体实施例,以及针对实施例的检测方法与结果。
实施例1实验于常温避光环境中执行
1、量取199mldi纯水备用;2、秤重取0.5g海藻酸;3、秤取0.068g硝酸银;4、0.2%(w/v)的30%过氧化氢溶液;5、将第2至4项反应物同时间加入前述纯水中;6、以高速搅拌机,转速700rpm搅拌5分钟,使第5项均匀混和;7、将第6项置于微波反应炉中,设定700w,时间10分钟;8、反应完成;9、将第8项迅速降温至40度c以下,终止反应,得到成品溶液;10、测得银晶体粒径为254nm。
实施例2实验于常温避光环境中执行
1、量取199mldi纯水备用;2、秤重取0.5g海藻酸;3、秤取0.068g硝酸银;4、0.2%(w/v)的30%过氧化氢溶液;5、将第2至4项反应物同时间加入前述纯水中;6、以高速搅拌机,转速700rpm搅拌5分钟,使第5项均匀混和;7、将第6项置于微波反应炉中,设定700w,时间20分钟。8、反应完成;9、将第8项迅速降温至40度c以下,终止反应,得到成品溶液;10、测得银晶体粒径为121.6nm。
实施例3实验于常温避光环境中执行
1、量取199mldi纯水备用;2、秤重取0.5g海藻酸;3、秤取0.068g硝酸银;4、0.1%(w/v)的30%过氧化氢溶液;5、将第2至4项反应物同时间加入前述纯水中;6、以高速搅拌机,转速700rpm搅拌5分钟,使第5项均匀混和;7、将第6项置于微波反应炉中,设定900w,时间20分钟;8、反应完成;9、将第8项迅速降温至40度c以下,终止反应,得到成品溶液;10、测得银晶体粒径为30nm。
对比例1实验于常温避光环境中执行
1、量取198mldi纯水备用;2、秤重取2.02g海藻酸;3、秤取0.068g硝酸银;4、0.1%(w/v)的30%过氧化氢溶液;5、将第2至4项反应物同时间加入前述纯水中;6、以高速搅拌机,转速700rpm搅拌5分钟,使第5项均匀混和;7、将第6项置于微波反应炉中,设定700w,时间10分钟;8、反应完成;9、将第8项迅速降温至40度c以下,终止反应,得到成品溶液;10、测得银晶体粒径大于仪器测定范围。
对比例2实验于常温避光环境中执行
1、量取198mldi纯水备用;2、秤重取2.02g海藻酸;3、秤取0.068g硝酸银;4、0.1%(w/v)的30%过氧化氢溶液;5、将第2至4项反应物同时间加入前述纯水中;6、以高速搅拌机,转速700rpm搅拌5分钟,使第5项均匀混和;7、将第6项静置8小时;8、将第7项置于微波反应炉中,设定700w,时间10分钟;9、得到未起反应的成品溶液。
对比例3实验于常温避光环境中执行
1、量取196.5mldi纯水备用;2、秤重取0.5g海藻酸;3、秤取0.068g硝酸银;4、0.5%(w/v)的还原剂葡萄糖;5、将第2至4项反应物同时间加入前述纯水中;6、以高速搅拌机,转速700rpm搅拌5分钟,使第5项均匀混和;7、将第7项置于微波反应炉中,设定700w,时间10分钟;8、反应完成;9、将第8项迅速降温至40度c以下,终止反应,得到成品溶液;10、成品溶液呈混浊情况,所得反应物非设计的结果。
效果实施例
测试项目与方法
1、金属晶体均匀性:将上述实施例中的含金属海藻酸单体结构以滤网过滤纯化后,使用zetasizernanozs90(malvern)机台进行测试。每一波峰代表单一金属晶体粒径,因此波峰数量代表金属粒径的数量,例如单一波峰代表溶液中的金属晶体具相同尺寸,二个波峰代表具有两个尺寸的金属晶体,以此类推。强度%代表金属晶体粒径所占比例,%比例越高,代表相同粒径越多,也同时代表金属晶体粒径尺寸的均匀性越高。
2、稳定度:依据上述金属晶体均匀性的测试方式,先对不同实施例分别进行测试得到金属经体粒径,之后置于在阴凉避光处6个月,再次测试金属晶体粒径,粒径变化率5%以下代表稳定。
3、抗菌效果:使用金黄色葡萄球菌(atcc出产的产品编号6538),日本标准jisl1902:2015菌液吸收法进行检测,抗菌活性值2.0以上代表抗菌。
4、细胞毒性:用水稀释成品溶液并以滤网过滤后,利用体外培养基扩散试验(invitroagardiffusiontest)进行检测,检测方式依据internationalorganization所发布的iso10993-5指引执行。(basedontheinternationalorganizationforstandardization(iso10993-5)guidelines),藉由细胞增生与凋亡的速率与数量判断是否具有细胞毒性。0代表无毒性,1:代表些微毒性,以此类推,数字越大表示毒性越强,最大数字为4。
测试结果:如下表1所示。
表1
上述对比例1中,由于所加入的海藻酸过多,导致银晶体粒径超过所需尺寸,测试结果表示难以(几乎无法)被细胞吸收,不符合本发明所需,因此未进行其余检测;对比例2中相较于对比例1多了静置8小时的步骤,明显超过本发明上述制程条件中所述的1小时,导致海藻酸单体被破坏,无法与金属离子进行反应从而得到含金属海藻酸单体结构;而对比例3中虽选用葡萄糖,但其相对于混合容易的重量百分浓度为0.5wt%,超过本发明限定的范围,结果得到的反应物并非设计结果,因此未进行后续检测。由表以及上述说明可得知,对比例1、2与3在各项度检测中的结果无法达到所需的标准;反观,本发明提供上述实施例1~3在金属晶体均匀性测试上都能有95%以上的均匀性,且其他测试项目的稳定度、抗菌效果以及细胞毒性也都有良好的表现,可以显示依据本发明提供的方法制作出的含金属海藻单体结构明显具有优良的结果。
以上所述,仅是本发明的较佳实施例而已,并非对本发明作任何形式上的限制,虽然本发明已以较佳实施例揭露如上,然而并非用以限定本发明,任何熟悉本专业的技术人员,在不脱离本发明技术方案范围内,当可利用上述揭示的方法及技术内容作出些许的更动或修饰为等同变化的等效实施例,但凡是未脱离本发明技术方案的内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与修饰,均仍属于本发明技术方案的范围内。
- 还没有人留言评论。精彩留言会获得点赞!