一种设计和构建猪全基因组CRISPR/Cas9敲除文库的方法与流程
本发明涉及生物
技术领域:
,具体地,涉及一种设计和构建猪全基因组crispr/cas9敲除文库的方法。
背景技术:
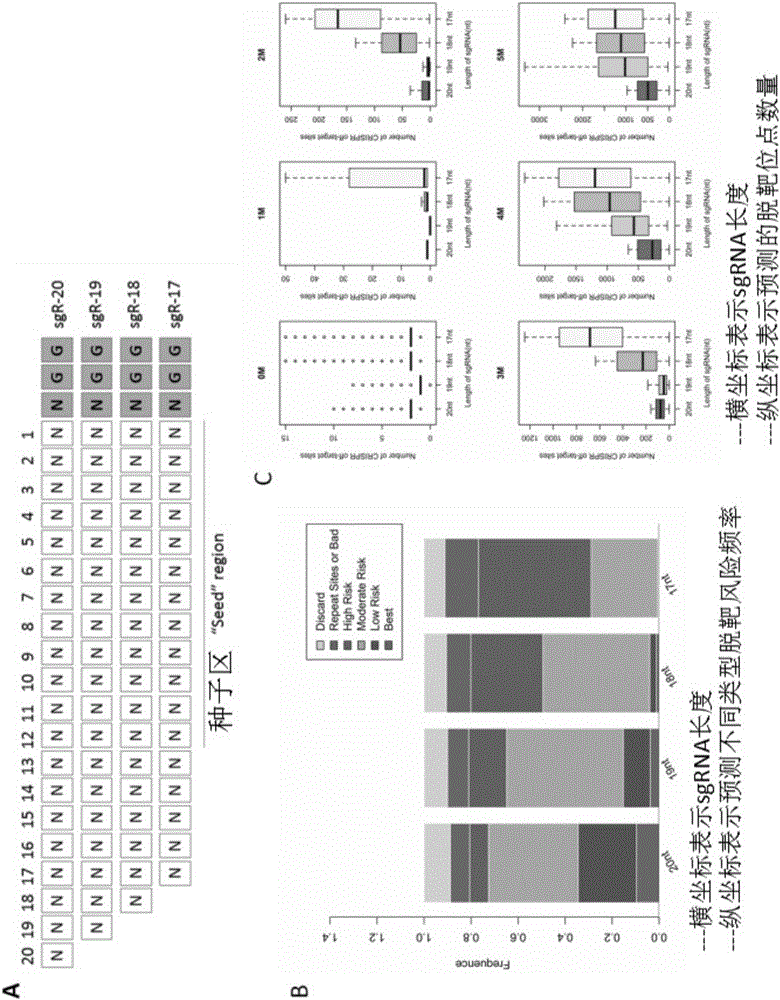
:利用功能缺失型(loss-of-function)或者功能获得型(gain-of-function)策略高通量筛选功能基因,是快速寻找调控特定表型的重要或关键基因的主要方法。传统采用rna干扰(rnainterference,rnai)文库进行遗传筛选方法因操作简单、成本相对较低等优势,尽管已经得到了广泛的应用,然而其抑制效果不完全、脱靶效应明显等劣势依然存在。近年来兴起的crispr/cas9技术能快速、简便、准确地实现基因组敲除等编辑功能,因而成为一种强大的遗传筛选工具。在各种细胞系、人和小鼠及斑马鱼等多种模式动物中,大规模运用该方法筛选功能基因已经取得了一些巨大成功。然而,目前猪的全基因组crispr/cas9敲除文库未见报道。因此,本领域迫切需要开发一种针对猪全基因组水平蛋白编码基因、lincrna和mirna设计sgrna,由此构建crispr/cas9的慢病毒敲除文库,即可利用该策略实现高通量筛选猪的功能基因的方法。技术实现要素:本发明的目的在于提供一种针对猪全基因组水平蛋白编码基因、lincrna和mirna设计sgrna,由此构建crispr/cas9的慢病毒敲除文库,即可利用该策略实现高通量筛选猪的功能基因的方法。本发明第一方面提供了一种猪全基因组特异性sgrna文库,所述文库包括:(i)n种表达猪特异性sgrna的载体,其中n为≥20000的正整数,所述的猪特异性sgrna是针对猪全基因组的靶基因,并且所述的靶基因选自下组:(a)蛋白编码基因、(b)lincrna基因、(c)mirna基因、(d)上述(a)、(b)和(c)的组合;并且,所述的猪特异性sgrna具有以下结构特征:(1)sgrna所针对的靶基因位点中含有的pam为ngg(n为a、t、c、g任意碱基);(2)sgrna长度为19或20nt;(3)sgrna的gc含量选择40-60%;(4)sgrna的全基因组脱靶评估选择不含1和2个碱基错配脱靶位点,且相比较其它sgrna,选择脱靶数量相对较少的sgrna;(5)对于靶向蛋白编码基因的猪特异性sgrna,其结合位置位于所述编码基因的起始密码子atg下游500bp区域内;对于靶向lincrna基因的猪特异性sgrna,其结合位置位于所述lincrna转录起始点下游的500bp区域内;对于靶向mirna基因的猪特异性sgrna,其结合位置为mirna前体序列或成熟序列;以及(ii)任选的m种阴性对照载体,所述的阴性对照载体为表达无关sgrna(无义sgrna)的载体,其中m为正整数。在另一优选例中,所述sgrna的序列特征为n20ngg(n为a、t、c、g任意碱基)。在另一优选例中,n为30000-150000,较佳地40000-100000,更佳地60000-90000。在另一优选例中,m为10-10000,较佳地50-5000,更佳地200-3000,最佳地500-2000。在另一优选例中,所述文库中,所述的n种猪特异性sgrna靶向≥90%(较佳地≥95%,更佳地96-100%或96-99%)的猪蛋白编码基因。在另一优选例中,所述文库中,所述的n种猪特异性sgrna靶向≥90%(较佳地≥95%,更佳地96-100%或96-99%)的猪lincrna基因。在另一优选例中,所述文库中,所述的n种猪特异性sgrna靶向≥90%(较佳地≥95%,更佳地96-100%或96-99%)的猪mirna基因。在另一优选例中,对于猪全基因组的全部基因而言,所述猪特异性sgrna的平均覆盖深度d1为2-10,较佳地2-6,更佳地为2-4(即,平均而言,每个基因有3种(或3条)针对该基因的猪特异性sgrna。在另一优选例中,所述的载体包括表达载体。在另一优选例中,所述的载体选自下组:质粒、病毒载体、或其组合。在另一优选例中,所述的病毒载体包括:慢病毒表达载体、腺病毒表达载体、转座子表达载体、或其组合。在另一优选例中,所述的表达载体除了含有表达cas9或sgrna的元件外,还含有表达慢病毒、腺病毒、转座子的元件。在另一优选例中,所述表达载体含有一表达猪sgrna的表达盒,所述表达盒具有式i所示的从5’至3’的结构:z0-z1-z2(i)式中,z0为第一启动子;z1为al-n20-ar,其中al为位于猪特异性sgrna的编码序列上游的左臂序列n20为猪特异性sgrna的编码序列,而ar为位于猪特异性sgrna的编码序列下游的右臂序列;z2为sgrnascaffold序列。在另一优选例中,所述表达盒具有式ii所示的从5’至3’的结构:z0-z1-z2-l-z3-z4(ii)式中,z0、z1、和z2如上所述,l为任选的间隔序列;z3为第二启动子;z4为报告基因。在另一优选例中,所述al为额外添加的含有限制性内切酶位点的正向引物结合序列。在另一优选例中,所述al为表达sgrna载体通过限制性内切酶线性化后,其紧邻限制性内切酶酶切位点上游的左侧同源载体序列。在另一优选例中,所述ar为额外添加的含有限制性内切酶位点的反向引物结合序列。在另一优选例中,所述ar为表达sgrna载体通过限制性内切酶线性化后,其紧邻限制性内切酶酶切位点下游的右侧同源载体序列。在另一优选例中,所述al的长度为25-40bp,较佳地25-35bp。在另一优选例中,所述n20的长度为20nt。在另一优选例中,所述ar的长度为25-40bp,较佳地28-35bp。在另一优选例中,所述第一启动子为启动sgrna表达的启动子。在另一优选例中,所述第一启动子选自下组:u6启动子、药物诱导型启动子(如teton/off系统)、或其组合。在另一优选例中,所述第二启动子选自下组:cmv启动子、cag启动子、cbh启动子、组织特异启动子、或其组合。在另一优选例中,所述报告基因选自下组:neomycin、puromycin、rfp、egfp、mcherry、blasticidin、或其组合。在另一优选例中,所述的表达载体选自下组:px330、lenti-sgrna-egfp、ad-sgrna、pb(piggybac)-sgrna、或其组合。在另一优选例中,所述的z1具有式(iv)结构:5’-tatcttgtggaaaggacgaaacaccgn20gttttagagctagaaatagcaagttaaaat-3’(iv)式中,n20为猪特异性sgrna的编码序列。在另一优选例中,所述sgrna文库的容量为82392条猪特异性sgrna。在另一优选例中,所述的文库是用本发明第二方面所述的方法制备的。本发明第二方面提供了一种制备本发明第一方面所述的猪全基因组特异性sgrna文库的方法,包括步骤:(a)提供核酸构建物z1,所述的核酸构建物z1具有以下结构:al-n20-ar其中,al为位于猪特异性sgrna的编码序列上游的左同源臂序列,n20为猪特异性sgrna的编码序列,ar为位于猪特异性sgrna的编码序列下游的右同源臂序列;(b)以所述的核酸构建物z1为模板,进行pcr扩增,从而获得扩增产物;(c)将所述扩增产物与经线性化的表达载体混合,并用gibsonassembly法进行连接,从而形成连接产物;(d)将所述连接产物转化大肠杆菌,从而获得转化子;(e)从所述转化子中抽提质粒,从而获得所述的猪全基因组特异性sgrna文库。在另一优选例中,所述的方法还包括:(f)将所述的质粒转入用于生产病毒的细胞,从而产生病毒颗粒(病毒载体)。在另一优选例中,所述的病毒包括慢病毒、和/或腺病毒。在另一优选例中,在步骤(a)中,所述核酸构建物z1为n1种,其中n1为≥20000的正整数。在另一优选例中,n1为30000-150000,较佳地40000-100000,更佳地60000-90000。在另一优选例中,所述的核酸构建物z1具有式(iv)结构。在另一优选例中,在步骤(c)中,所述的pcr扩增所用的引物为:f:5’-ggctttatatatcttgtggaaaggacgaaacaccg-3’(seqidno.:130);r:5’-ctagccttattttaacttgctatttctagctctaaaac-3’(seqidno.:131)。在另一优选例中,在步骤(c)中,所述的线性化表达载体是用bbsi酶切产生的。在另一优选例中,所述的表达载体选自下组:px330、lenti-sgrna-egfp、ad-sgrna、pb(piggybac)-sgrna、或其组合。在另一优选例中,所述的表达载体为lenti-sgrna-egfp。在另一优选例中,所述方法还包括:(g)在低moi条件(例如0.1-0.8,较佳地0.2-0.4)下,将所述病毒载体感染猪细胞。在另一优选例中,在步骤(g)中,每个猪细胞基本被1个病毒载体(或病毒粒子)感染。在另一优选例中,所述的猪细胞选自下组:猪pk15细胞、3d4/21细胞、或其组合。本发明第三方面提供了一种试剂盒,所述试剂盒包括:(a)第一容器以及位于所述容器中的如本发明第一方面所述的猪全基因组特异性sgrna文库;以及(b)使用说明书,所述的使用说明书记载了所述猪全基因组特异性sgrna文库的使用说明。在另一优选例中,所述的试剂盒还包括:(c)第二容器以及位于所述第二容器中的猪细胞,所述的猪细胞含有组成型或诱导型表达cas9蛋白。在另一优选例中,所述的猪细胞选自下组:猪pk15细胞、3d4/21细胞、或其组合。本发明第四方面提供了一种如本发明第一方面所述的sgrna文库的用途,用于制备高通量筛选猪的功能基因的试剂盒,或用于筛选猪的功能基因。在另一优选例中,所述功能基因选自下组:蛋白编码基因、lincrna、mirna、或其组合。在另一优选例中,所述的筛选是非治疗性和非诊断性的方法。应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例)中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。限于篇幅,在此不再一一累述。附图说明图1是本发明实施例1中对长度为17nt、18nt、19nt和20nt的sgrna进行脱靶风险评估。a.不同长度sgrna示意图;b.利用sgrnacas9软件针对100个基因设计sgrna并比较不同长度sgrna的脱靶分析;c.不同长度sgrna的脱靶位点错配碱基比较。图2是本发明实施例1中针对7个蛋白编码基因和2个mirna靶向同一位点不同长度sgrna脱靶位点数量对比分析。图中,0m表示sgrna打靶位点,1m-5m表示sgrna脱靶位点。其中,1m表示含有一个碱基错配的脱靶位点;2m表示含有两个碱基错配的脱靶位点;3m表示含有三个碱基错配的脱靶位点;4m表示含有4个碱基错配的脱靶位点;5m表示含有5个碱基错配的脱靶位点,totalno.ofot表示总的脱靶位点数量。图3是本发明实施例1中针对7个蛋白编码基因和2个mirna靶向同一位点不同长度sgrna脱靶位点韦恩图分析。图4是本发明实施例1中针对7个蛋白编码基因和2个mirna靶向同一位点不同长度sgrna活性t7en1酶切鉴定。图5是本发明实施例1中针对靶向ar基因随机挑选的19个脱靶位点验证。图6是本发明实施例3中方案(1)t4dna连接酶法实验示意图。图7是本发明实施例3中猪全基因组sgrna合成的寡核苷酸池高通量测序分析。图8是本发明实施例3中比较用于构建文库的pcr产物含量对连接后克隆数生长的影响。图9是本发明实施例3中方案(2)gibsonassembly法实验示意图。图10是本发明实施例3中比较一个转化反应用不同量gibson组装产物对克隆生长的影响。图11是本发明实施例5中比较一个连接反应用不同文库扩增纯化产物量对克隆生长的影响。图12是本发明实施例6中筛选特异性扩增猪全基因组sgrna质粒文库目的片段最优引物对。图13是本发明实施例7中构建稳定表达cas9的pk-15细胞系及其活性鉴定。图14是本发明实施例8中慢病毒载体表达sgrna活性测试,a图用评估gfp荧光蛋白表达量表示sgrna慢病毒载体活性高低,b图为检测的靶向anpep基因的sgrna慢病毒载体为例说明文库的质粒系统能有效切割基因组靶点。图15是本发明实施例9中慢病毒文库感染筛选突变细胞株测试,*表示有基因组编辑活性的sgrna靶点,其中pcr扩增结果表示有大片段缺失突变。图16是猪的全基因组crispr/cas9敲除文库设计案例。具体实施方式本发明人通过广泛而深入的研究,首次开发一种针对猪全基因组水平蛋白编码基因、lincrna和mirna设计sgrna,由此构建crispr/cas9的慢病毒敲除文库,可利用该策略实现高通量筛选猪的功能基因的方法。在此基础上,本发明人完成了本发明。具体地,本发明开发了一个猪的全基因组crispr/cas9敲除文库质粒试剂盒,包括cas9表达载体和82392个sgrna表达载体库,分别靶向猪的14958个编码基因,14168个lincrna和142个mirna,并含有970个阴性对照sgrna,同时还构建了一种稳定表达cas9的pk-15细胞株,两者配合使用,可用于全基因组水平高通量筛选猪的功能基因。术语如本文所用,术语“质粒”也可以被称为表达载体,当其被转入细胞中时,可以表达其中所包含的与启动子相连的编码序列或非编码序列。质粒中通常含有基因表达所必需的元件。在本发明中,所使用的质粒可以是真核基因表达质粒、慢病毒载体质粒,或者任何其他可以表达其中所含编码或非编码基因的质粒,优选为慢病毒载体质粒。如本文所用,术语“gibson组装”又称为gibson由j.craigventer研究所的danielgibson博士在2009年提出。gibson组装方法适合于拼接多个线性dna片段,也适合将目的dna插入载体中。进行gibson组装时,首先需要在dna片段的末端加上同源片段,然后将这些dna片段和mastermix混合孵育1小时,mastermix由三种酶组成,其中dna外切酶先从5’端降解核苷酸产生粘性末端,然后相邻片段之间的重叠序列退火,最后dna聚合酶和dna连接酶将序列填充,形成完整的双链dna分子,实现无痕拼接。mastermix可以从neb或sgi-dna公司购买,或者自己配制(参见millerlabprotocol)。如本文所用,术语“报告基因”、“标记物基因”可互换使用,指其表达可以被选择或富集的任何标记物基因,即当该标记物基因在细胞中表达时,可以通过一定方式选择和富集表达该标记物基因的细胞。可用于本发明的报告基因包括但不限于在表达后可以用facs分选的荧光蛋白基因,或者可以利用抗生素进行筛选的抗性基因,或者表达后可以被对应抗体识别并通过免疫染色或磁珠吸附进行筛选的蛋白基因。可用于本发明的抗性基因包括但不限于针对杀稻瘟菌素(blasticidin)、遗传霉素(geneticin,g-418)、潮霉素(hygromycinb)、霉盼酸(mycophenolicacid)、瞟岭霉素(puromycin)、博莱霉素(zeocin)或新霉素(neomycin)的抗性基因。可用于本发明的荧光蛋白基因包括但不限于蓝色荧光蛋白(cyanfluorescentprotein)、绿色荧光蛋白(greenfluorescentprotein)、增强绿色荧光蛋白(enhancedgreenfluorescentprotein)、黄色荧光蛋白(yellowfluorescentprotein)、橙色荧光蛋白(orangefluorescentprotein)、红色荧光蛋白(redfluorescentprotein)、远红色荧光蛋白(far-redfluorescentprotein)或可切换荧光蛋白(switchablefluorescentproteins)的基因,优选为增强绿色荧光蛋白(egfp)。sgrna通常,sgrna又称为“单链向导rna”,是crrna和trancrrna融合而成的一条rna链,通常,如本领域技术人员所熟知的,sgrna包含与靶序列配对的序列(也被称为grna配对序列或sgrna配对序列)、骨架(scaffold)序列(也被称为grna骨架序列或sgrna骨架序列)和转录终止子(例如polya)。在本发明中,除非特别说明,grna和sgrna可以互换使用。在本发明中,术语“grna骨架序列”或“sgrna骨架序列”是指sgrna中grna与靶标基因组配对序列与转录终止子之间的序列。在一优选实施方式中,所述sgrna骨架序列选自下组:靶标位点配对序列,sgrnascaffold和转录终止序列“tttttt”,具体的序列为序列为:gttttagagctagaaatagcaagttaaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgctttttt,其中,划横线部分为检测引物-r。在本发明中,grna(sgrna)的设计方法和设计工具可以是常规的,可通过多种途径获得,包括但不限于:sgrnacas9、e-crisp和/或chopchop。猪全基因组特异性sgrna文库本发明提供了一种猪全基因组特异性sgrna文库,所述文库包括:(i)n种表达猪特异性sgrna的载体,其中n为≥20000的正整数,所述的猪特异性sgrna是针对猪全基因组的靶基因,并且所述的靶基因选自下组:(a)蛋白编码基因、(b)lincrna基因、(c)mirna基因、(d)上述(a)、(b)和(c)的组合;并且,所述的猪特异性sgrna具有以下结构特征:(1)sgrna所针对的靶基因位点含有的pam为ngg,优选地,sgrna为n20ngg(n为a、t、c、g任意碱基);(2)sgrna长度为19或20nt;(3)sgrna的gc含量选择40-60%;(4)sgrna的全基因组脱靶评估选择不含1和2个碱基错配脱靶位点,且相比较其它sgrna,选择脱靶数量相对最少的sgrna;(5)对于靶向蛋白编码基因的猪特异性sgrna,其结合位置位于所述编码基因的起始密码子atg下游500bp区域内;对于靶向lincrna基因的猪特异性sgrna,其结合位置位于所述lincrna转录起始点下游的500bp区域内对于靶向mirna基因的猪特异性sgrna,其结合位置为mirna前体序列或成熟序列;以及(ii)任选的m种阴性对照载体,所述的阴性对照载体为表达无关sgrna的载体,其中m为正整数。在一优选实施方式中,一种全基因组crispr/cas9敲除文库设计方法如下所示:使用的软件为sgrnacas9,网址为:http://biootools.com/col.jsp?id=143,版本号为“sgrnacas9_3.0.5”。主要技术指标依据实验数据确定最佳参数,包括:sgrna识别基因组上的pam可选nag或ngg,优选ngg;sgrna长度可选范围为17nt、18nt、19nt和20nt,优选20nt;sgrna的gc含量可选范围为20%-80%,优选40%-60%;sgrna的全基因组脱靶评估考察5个碱基错配,优选基因组上唯一存在,不含1和2个碱基错配脱靶位点,且全部脱靶位点最少的sgrna。对于sgrna定位,靶向编码基因全长序列,即cds编码区,优选起始密码子atg下游500bp区域内;靶向lincrna全长序列,优选转录起始500bp区域内;靶向mirna全长序列,优选靶向mirna前体序列,更优选靶向mirna的成熟序列。一个基因可设计1~10条sgrna,优选一个基因设计3~5条sgrna;sgrna文库需设计无义sgrna作为阴性对照,作为阴性对照的sgrna在基因组上无靶点,可选100~2000条,优选设计1000条。构建文库的方法本发明提供了一种制备本发明所述的猪全基因组特异性sgrna文库的方法,包括步骤:(a)提供核酸构建物z1,所述的核酸构建物z1具有以下结构:al-n20-ar其中,al为位于猪特异性sgrna的编码序列上游的左同源臂序列(优选地,表达sgrna载体通过限制性内切酶线性化后,其紧邻限制性内切酶酶切位点上游的左侧同源载体序列),n20为猪特异性sgrna的编码序列,ar为位于猪特异性sgrna的编码序列下游的右同源臂序列(优选地,表达sgrna载体通过限制性内切酶线性化后,其紧邻限制性内切酶酶切位点下游的右侧同源载体序列);(b)以所述的核酸构建物z1为模板,进行pcr扩增,从而获得扩增产物;(c)将所述扩增产物与经线性化的表达载体混合,并用gibsonassembly法进行连接,从而形成连接产物;(d)将所述连接产物转化大肠杆菌,从而获得转化子;(e)从所述转化子中抽提质粒,从而获得所述的猪全基因组特异性sgrna文库。在一优选实施方式中,一种全基因组crispr/cas9敲除文库构建方法如下所示:(1)可选的载体骨架有普通sgrna表达载体,如px330、px458,sgrna慢病毒表达载体,如lenti-sgrna-egfp。为确保sgrna文库中绝大多数sgrna进入细胞,优选sgrna慢病毒表达载体(lenti-sgrna-egfp),即通过慢病毒感染细胞,以达到较高的整合效率。其载体可携带筛选标记基因,如neo、puro、bfp、egfp,优选egfp,可方便后续通过流式细胞仪分选阳性感染细胞。(2)将合成的包含sgrna序列的寡核苷酸探针池连接进sgrna慢病毒表达载体,可选的方法有:(a)sgrna序列两侧连接pcr扩增的接头序列:5’-acaggcccaagatcgtgaagaagacagcaccgn20gtttctgtcttctccaagaagcgcaaggag-3’(n20代表sgrna序列),用高通量的方法合成85674条寡核苷酸探针池,通过pcr扩增(扩增引物序列为f:5’-acaggcccaagatcgtgaa-3’(seqidno.:127),r:5’-ctccttgcgcttcttgga-3’(seqidno.:128))、酶切和连接到目标载体,挑选单克隆菌落sanger测序鉴定连接效率;(b)sgrna序列两侧附加同源臂,同源臂为目标载体酶切线性化后两端序列:5’-tatcttgtggaaaggacgaaacaccgn20gttttagagctagaaatagcaagttaaaat-3’(n20为sgrna序列),用高通量方法合成85674条寡核苷酸探针池,然后用pcr扩增(扩增引物序列为:f:5’-ggctttatatatcttgtggaaaggacgaaacaccg-3’(seqidno.:130,r:5’-ctagccttattttaacttgctatttctagctctaaaac-3’(seqidno.:131))、gibsonassembly方法与目标载体通过同源重组连接。先通过ta克隆测序法鉴定连接效率,最后通过高通量测序法鉴定文库中sgrna序列丰度。比较两种方法构建慢病毒表达sgrna载体的连接效率,结果表明:方法b>方法a。(3)将连接成功的慢病毒表达sgrna载体转化到大肠杆菌细胞中,扩大培养和提取质粒,可选:1)化学转化,2)电转化,由于国内很难购买高质量的电转化感受态细胞,如neb公司,thermo公司等。所以优选化学转化法,所采用的大肠杆菌为trans1-t1phageresistantchemicallycompetentcell(transgen),再使用gibsonassembly组装策略,可达到较高的转化效率,可获得转化子数量覆盖度为200x的sgrnas重组载体库。(4)转化后lb平板上的克隆菌株,可通过刮取到液体培养基中扩大培养,或转化后直接接种到液体培养基中扩大培养,置于37℃摇床12-16h后,收集细菌提取质粒。由于化学转化相比电转化效率较低,需要大量的细菌转化反应,涂平板刮取克隆的方法工作量巨大,故优选转化后直接接种的方法。(5)鉴定crispr/cas9质粒文库sgrna序列的覆盖度和丰度。利用pcr扩增、建库和高通量测序的方法。为了达到最佳检测效果,优选引物对组合为lenti-f1/r6(121bp)或lenti-f3/r6(153bp),如下:(1)lenti-f1:5’-cttgtggaaaggacgaaacacc-3’,seqidno.:132,lenti-r6:5’-aagcaccgactcggtgcca-3’,seqidno.:140;(2)lenti-f3:5’-gaaagtatttcgatttcttggc-3’,seqidno.:134,lenti-r6:5’-aagcaccgactcggtgcca-3’,seqidno.:140。(6)测试crispr/cas9质粒文库慢病毒包装和感染能力,利用无内毒素质粒大抽试剂盒提取sgrna混池质粒,在hek293t细胞中进行慢病毒包装,收集病毒原液,离心浓缩,测定滴度,取一定量病毒液感染hek293t和pk-15细胞,即可依据gfp亮度强弱判定慢病毒文库是否可使用。混合质粒包装慢病毒效率要低于单个质粒,需大量慢病毒sgrna质粒转染293t细胞,以达到较高的病毒滴度。(7)将sgrna慢病毒文库感染稳定表达cas9的pk-15细胞系中,并挑取表达gfp的单克隆细胞。根据测序结果确定稳定整合细胞中的sgrna,并验证sgrna相对应的基因是否被编辑。试剂盒本发明提供了一种试剂盒,所述试剂盒包括:(a)第一容器以及位于所述容器中的如本发明所述的猪全基因组特异性sgrna文库表达载体;以及(b)使用说明书,所述的使用说明书记载了所述猪全基因组特异性sgrna文库的使用说明。在另一优选例中,所述的试剂盒还包括:(c)第二容器以及位于所述第二容器中的猪细胞,所述的猪细胞组成型或诱导型cas9蛋白表达载体。在另一优选例中,所述的猪细胞选自下组:猪pk15细胞、3d4/21细胞、或其组合。在一优选实施方式中,所述猪细胞为能稳定表达cas9的pk-15细胞株。利用该细胞株与猪的全基因组crispr/cas9敲除文库质粒试剂盒配合使用,可提高基因组敲除效率,实现高通量筛选猪的功能基因。在一优选实施方式中,本发明提供了一种猪的全基因组crispr/cas9敲除文库质粒试剂盒。所述试剂盒包括表达cas9的慢病毒载体和通过本发明的方法设计和构建的猪的全基因组sgrna慢病毒质粒,通过高通量测序鉴定发现共获得了82392条sgrna,可分别靶向猪的14958个编码基因,14168个lincrna和142个mirna,并含有970个阴性对照sgrna。本发明的主要优点包括:(1)本发明首次公开了一种全基因组crispr/cas9敲除文库设计和构建方法,全基因组水平设计sgrna周期短,合成的sgrna寡核苷酸序列准确性高,文库构建过程中sgrna重组载体连接效率高。(2)本发明首次公开了一种猪的全基因组crispr/cas9敲除文库质粒试剂盒,该试剂盒包含的sgrna特异性高,靶标位点覆盖猪的全基因组编码或非编码基因,非常适用于高通量筛选猪的功能基因。(3)本发明还提供了一种稳定表达cas9的pk-15细胞株,使用该细胞株可减少转染外源cas9表达载体,同时提高基因组编辑效率。下面结合具体实施例,进一步陈述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明详细条件的实验方法,通常按照常规条件如sambrook等人,分子克隆:实验室手册(newyork:coldspringharborlaboratorypress,1989)中所述的条件,或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数按重量计算。除非有特别说明,否则实施例所用的材料均为市售产品。实施例1基于实验数据确定最佳参数用于全基因组crispr/cas9敲除文库设计方案利用全基因组crispr/cas9敲除文库可开展高通量筛选功能基因研究。但crispr/cas9系统存在脱靶效应,其次是不同长度的sgrna活性或脱靶效应可能不同。为了确定最佳的全基因组crispr/cas9敲除文库设计参数。利用sgrnacas9软件,针对人基因组中随机选择的100个基因设计sgrna和评估脱靶效应,同时比较长度为17nt、18nt、19nt和20nt的sgrna脱靶效应,以及实验验证四种不同长度sgrna的活性和特异性规律。由图1a可见,选择的pam为ngg;对于sgrna的特异性评估或脱靶效应分类分别为弃用(discard):在基因组上无对应的靶标结合位点;高风险(high_risk):存在1个碱基错配的脱靶位点,其中含种子序列完全匹配,且非种子序列仅含1个碱基错配的脱靶位点;中等风险(moderate_risk):存在2个碱基错配的脱靶位点,其中含种子序列完全匹配,且非种子序列含不多于2个碱基错配的脱靶位点;低风险(low_risk):存在多于2个碱基错配的脱靶位点;识别位点重复或较差(repeat_sites_or_bad):存在多个完全匹配的靶标结合位点;最佳(best):不存在少于2个碱基错配的脱靶位点,且其中不含种子序列完全匹配的脱靶结合位点。由图1b可见,随着sgrna长度变短,最佳(best)的sgrna的数量越来越少,而highrisk(高风险)的sgrna的数量越来越多。同时从图1c可见,引起这种变化的原因是随着sgrna变短,预测的脱靶位点含有1或2个碱基错配的数目增加,而这就可能增加脱靶风险。此外,我们随机选择了靶向7个基因和2个mirna同一位点不同长度的sgrna进行脱靶比较研究,由图2可见,发现sgrna预测到的脱靶位点数量随着长度变短显著增加。通过韦恩图分析,发现长度为17nt的sgrna预测的脱靶位点数量最多,且包含18nt,19nt和20nt共有脱靶位点(图3)。进一步,我们在hek293t细胞上,对不同长度sgrna活性和脱靶效应进行了验证。其中靶向目标基因的sgrna序列和引物序列如下所示:sgrna序列为:用于构建sgrna表达载体的引物为:设计的引物送公司合成,退火后与bbsi消化的px330(全称px330-u6-chimeric_bb-cbh-hspcas9,addgene#42230)载体连接,sgrna引物退火体系(10μl):sgrna-f:5μl和sgrna-r:5μl,退火程序:95℃,10min;65℃,1h。通过bbsi酶切位点将退火产物连接进酶切后的px330载体中,通过测序鉴定成功后,利用无内毒素质粒提取试剂盒(omega,d6950-01)抽提质粒,nandrop2000浓度仪测定质粒浓度,随后转染至汇合度约80%的hek293t细胞,按照转染试剂lipofectamine2000(thermo,#11668027)说明书转染细胞。我们对靶向7个基因和2个mirna同一位点不同长度sgrna进行切割活性比较研究,转染48h后收集细胞,按照天根dna提取试剂盒(tiangen,dp304-03)说明书提取dna,通过pcr扩增,pcr扩增引物如下:dmd-ftcacccaatgccagtgagatseqidno.:109dmd-rtatctccaggagcacagccaseqidno.:110esr-fctggatccgtctttcgcgttseqidno.:111esr-rgctcgttctccaggtagtaggseqidno.:112mir-21-ftgaaactgggctgctgcataseqidno.:113mir-21-raaccacgactagaggctgacseqidno.:114ar-fgccgtccaagacctaccgaseqidno.:115ar-ratgctccaacgcctccacaseqidno.:116tp53-faagggagttgggaatagggtgseqidno.:117tp53-ragggggactgtagatgggtgaseqidno.:118crygc-fggcattctacctcagcaaccseqidno.:119crygc-raacctccctccctgtaaccseqidno.:120mstn-fagattcactggtgtggcaagseqidno.:121mstn-rgattgtttccgttgtagcgtgseqidno.:122mir-206a-fgagtggctctctgcgtgaatseqidno.:123mir-206a-rgcttccttggtgagggagtcseqidno.:124igf2-ftaacacggctctctctgtgcseqidno.:125igf2-rgagtagcctgtttcggggagseqidno.:126pcr反应体系为:10xlapcrbuffer:5μl;dntpmixture(mg2+plus):4μl;前向引物:1μl;反向引物:1μl;dna:(20~50)ng;补水至50μl。pcr反应程序为:95℃,10min;32个循环(95℃,30s;60℃,30s;72℃,1min);72℃,5min;15℃,2min。pcr完成后按照takara纯化试剂盒(takara,9701)说明书纯化pcr产物,在pcr仪上进行退火反应,退火反应总体积为(19.5μl):10xnebuffer:2μl,,dna:200ng,补水至19.5μl。退火程序为:95℃,5min;95-85℃,-2℃/s;85-25℃,-0.1℃/s;4℃,2min。退火后用t7en1(neb,m0302)酶切,酶切体系为:退火后的pcr产物:19.5μl,t7endonucleasei:0.5μl。37℃水浴酶切15min,用2%的普通琼脂糖凝胶电泳检测。结果如图4所示。不同长度sgrna活性高低与靶点有关,如靶向dmd基因,长度为18nt的sgrna切割活性最高,而靶向esr基因的长度为17nt和18nt的sgrna无活性,靶向mir-21的不同长度sgrna均有活性,且活性相当。同时,针对ar基因不同长度sgrna随机选择19个位点进行脱靶验证,采用pcr产物直接测序和t7en1酶切,通过pcr测序结果通过tide软件(https://tide.nki.nl/)评估效率,由图5(a,b)可见,不同长度sgrna的脱靶位点含有1-5个错配碱基,均存在基因组切割,设定切割效率大于6%的位点具有脱靶活性,不同长度能被验证的脱靶位点数量相当,说明不同长度sgrna均存在脱靶效应,但从预测到的脱靶数量显示,短的sgrna预测的脱靶数量显著增加。因此,优先考虑长度为20nt的sgrna。基于以上实验数据和公开的文献报道,最终确定设计特异性高的全基因组crispr/cas9敲除文库最佳参数如下:(1)sgrna的pam选择ngg(优选地,sgrna为n20ngg(n为a、t、c、g任意碱基));(2)sgrna长度为19或20nt;(3)sgrna的gc含量选择40-60%;(4)sgrna的全基因组脱靶评估选择不含1和2个碱基错配脱靶位点,且相比较其它sgrna,选择脱靶数量相对较少的sgrna;(5)对于sgrna定位,若靶向编码基因选择起始密码子atg下游500bp区域内;若靶向lincrna全长序列,选择转录起始靠前500bp区域内;若靶向mirna全长序列,选择靶向mirna前体序列或成熟序列。(6)每个基因设计3条sgrna;(7)sgrna文库所需的阴性对照,即无义sgrna,选择1000条。实施例2设计猪的全基因组crispr/cas9敲除文库以猪的所有编码蛋白基因,mirna和lincrna为研究对象,按照方案1中确定的最佳参数设计猪的全基因组crispr/cas9敲除文库,其中编码蛋白基因和基因组序列从为ensembl数据库下载:ftp://ftp.ensembl.org/pub/release-87/fasta/sus_scrofa/cds/和ftp://ftp.ensembl.org/pub/release-87/fasta/sus_scrofa/dna/。猪的mirna序列从mirbase数据库下载:http://www.mirbase.org/。lincrna的序列从aldb数据库下载:http://res.xaut.edu.cn/aldb/download.jsp。sgrna所针对的靶基因位点含有的pam为ngg,sgrna长度为20nt,gc含量范围为40%-60%,sgrna的全基因组脱靶评估考察5个碱基错配,优选基因组上唯一存在,不含1和2个碱基错配脱靶位点,且全部脱靶位点最少的sgrna。一个基因设计3条sgrna;设计1000条在基因组上无靶点的sgrna作为阴性对照。总共设计有85674条sgrna,分别靶向17743个编码蛋白基因,11053个lincrna和551个mirna。基于设计好的针对猪基因组的sgrna,采用人工合成方式制得式iii或式iv结构的核酸构建物:5’-acaggcccaagatcgtgaagaagacagcaccgn20gtttctgtcttctccaagaagcgcaaggag-3’(iii)5’-tatcttgtggaaaggacgaaacaccgn20gttttagagctagaaatagcaagttaaaat-3’(iv)式中,n20为sgrna序列。实施例3比较两种不同策略构建猪sgrna文库的效率方案(1)t4dna连接酶法sgrna序列两侧连接pcr扩增的接头序列,含bbsi酶切位点,设计的接头序列如下:5’-acaggcccaagatcgtgaagaagacagcaccgn20gtttctgtcttctccaagaagcgcaaggag-3’(n20为sgrna序列),pcr产物大小为:82bp,其中t4dna连接酶法文库构建流程见图6。bbsin205-acaggcccaagatcgtgaagaagacagcaccgnnnnnnnnnnnnnnnnnnnngtttctgtcttctccaagaagcgcaaggag-33-tgtccgggttctagcacttcttctgtcgtggcnnnnnnnnnnnnnnnnnnnncaaagacagaagaggttcttcgcgttcctc-5bbsi探针设计案例:>enssscg00000000001_5_596292_596321_pacaggcccaagatcgtgaagaagacagcaccgcacgacgtagtagaagcgcagtttctgtcttctccaagaagcgcaaggag(下划线部分为sgrna序列)将长度为82mer的序列送美国customarrayinc,合成85674条寡核苷酸池,随后对合成的寡核苷酸池进行文库构建和高通量测序鉴定,结果如图7所示,准确率为99.23%。接着以寡核苷酸池为模板,进行pcr扩增,其中扩增引物序列为:前向引物f:5’-acaggcccaagatcgtgaa-3’(seqidno.:127),反向引物r:5’-ctccttgcgcttcttgga-3’(seqidno.:128),pcr反应体系为:2xphusionhigh-fidelitypcrmastermixwithhfbuffer:25μl;sgrnalibrary:1μl;前向引物f:2μl;反向引物r:2μl;灭菌水:20μl。反应程序为:98℃,2min;分别设置8,10,12或16个循环(98℃,10s;60℃,15s;72℃,45s);72℃,5min;15℃,2min。用4%琼脂糖凝胶电泳,通过照胶仪观察目的条带位置。切下目的条带(82bp),根据qiagengelextractionkit(qiagen,28704)说明书回收目的条带,最后用10μl洗脱;利用bbsi限制性内切酶酶切纯化后的pcr产物,酶切反应体系为:10xfastdigestbuffer:3μl;回收后pcr产物:400ng;bbsi:2μl;补灭菌水至30μl。酶切lenti-sgrna-egfp载体,酶切反应体系为:10xfastdigestbuffer:2μl;lenti-sgrna-gfp:3μg;bbsi:3μl;补灭菌水至20μl。置于37℃水浴酶切3h以上,酶切产物用20%丙烯酰胺凝胶电泳,电泳液使用0.5xtbe,上样缓冲液用rnaloadingbuffer,调至100v电压在冰上电泳。20%丙烯酰胺凝胶配制:(29%丙烯酰胺和1%n,n’-亚甲基双丙烯酰胺)29:1:13.32ml;灭菌水:2.54ml;5xtbe:4ml;10%过硫酸铵(ap):0.14ml;temed:5μl。从page凝胶中回收dna片段:(1)在凝胶尚未完全干燥时用洁净的手术刀片切取目的dna条带,放入干净的1.5ml离心管中,加500μl双蒸水洗涤,离心并吸弃双蒸水;(2)再加入500μl双蒸水,用枪头尖将凝胶碾碎,37℃水浴12h;(3)12000rpm离心5min后,将上清液(dna粗回收液)移至另一新的干净1.5ml离心管中;(4)于上清液(dna粗回收液)中加入2倍体积预冷无水乙醇和1/10体积3m醋酸钠(ph=5.2),在-20℃冰箱中放置1h;(5)4℃,12000rpm离心20min,吸弃上清;(6)用75%乙醇洗涤沉淀2次,4℃,1200rpm离心2min,吸弃上清;(7)待乙醇挥发干净后,用双蒸水溶解沉淀;(8)用nanodrop2000检测回收后产物浓度。再按照连接酶(takara,6023)说明书进行连接。利用该方案,检测了利用1ng,5ng,10ng和50ng纯化的pcr产物进行连接转化,统计克隆总数,如图8所示,生长的克隆数为:0.37x103~3x103个。此外,随机挑选50个单克隆菌落扩大培养,送公司测序,测序引物为:5’-gcatatacgatacaaggctg-3’(seqidno.:129),获得的测序阳性菌落为38个,阳性率为76%。方案(2)gibsonassembly法sgrna序列两侧加同源臂,同源臂为目标载体线性化后两端序列,同源臂序列设计为:5’-tatcttgtggaaaggacgaaacaccgn20gttttagagctagaaatagcaagttaaaat-3’(n20为sgrna序列),其中gibsonassembly法文库构建流程见图9,同样送美国customarrayinc公司合成85674条寡核苷酸池。探针示例为:>enssscg00000000001_5_592106_592135_mtatcttgtggaaaggacgaaacaccgtgacgactcggccaccaccagttttagagctagaaatagcaagttaaaat(下划线为sgrna序列)随后对合成的寡核苷酸池进行文库构建。其中,利用寡核苷酸池为模板进行pcr扩增,其中扩增的引物序列为:f:5’-ggctttatatatcttgtggaaaggacgaaacaccg-3’(seqidno.:130);r:5’-ctagccttattttaacttgctatttctagctctaaaac-3’(seqidno.:131),将获得的pcr扩增产物,按照gibsonassembly(neb,e2611l)试剂盒说明书方法与目标载体同源重组连接,组装产物转化到大肠杆菌细胞中,涂板并培养。对于获得的转化子,在提取质粒后,通过高通量测序进行鉴定。实施例4评估不同量的gibson组装液对转化效率的影响pcr纯化产物量为60ng,酶切后的lenti-sgrna-gfp载体量为100ng。一个转化反应所用大肠杆菌量为50μl,gibson组装产物量分别为5μl、10μl、20μl,最后统计克隆数(gibson组装产物是实施例3中的组装产物,统计结果由图10所示,当所用gibson组装产物量为5μl时,可达到较高的转化效率,此时一个连接反应中大肠杆菌的数量约为0.5x109个。实施例5评估不同量的文库扩增纯化产物对转化效率的影响在本实施例中,检测利用5ng~450ngpcr扩增产物,进行连接转化获得的克隆,统计克隆数,从而评估不同量的文库扩增纯化产物对转化效率的影响。如图11所示,生长的克隆数为:3.5x103至10x104个。由此可见,采用实施例3的方案2获得的重组克隆数高于实施例3的方案1。同样通过随机挑选50个单菌落扩大培养送公司测序,获得的阳性菌落为48个,阳性率为96%,高于方案1。统计结果显示,当一个转化反应用文库扩增纯化产物量为10ng,gibson组装产物量为5μl时,可达到非常高的转化效率,最终大量转化可获得转化子数量覆盖度为200x的sgrnas重组载体库。进一步,通过构建文库和高通量测序法来鉴定文库中sgrna的覆盖度。实施例6筛选特异性扩增猪全基因组sgrna质粒文库目的片段最优引物对利用实施例3中方案(2)gibsonassembly法构建猪的全基因组sgrna质粒文库后,为了确定其中连接的sgrna种类和丰度,以质粒文库为模板,先通过pcr扩增含有sgrna序列的目标区域,其中sgrna表达载体骨架为lenti-sgrna-egfp,共设计了3条正向引物(lenti-f1:cttgtggaaaggacgaaacacc(seqidno.:132);lenti-f2:atatcttgtggaaaggacg(seqidno.:133);lenti-f3:gaaagtatttcgatttcttggc(seqidno.:134)),7条反向引物(lenti-r1:caagttgataacggactagcc(seqidno.:135);lenti-r2:cagaattggcgcacgcgctaa(seqidno.:136);lenti-r3:gagccatttgtctgcagaattggc(seqidno.:137);lenti-r4:ggctagcggtacctctagagcc(seqidno.:138);lenti-r5:ccacaggctagcggtacctct(seqidno.:139);lenti-r6:aagcaccgactcggtgcca(seqidno.:140);lenti-r7:cttgctatttctagctct(seqidno.:141))。分别组合(lenti-f1/r1、lenti-f1/r2、lenti-f1/r3、lenti-f1/r4、lenti-f1/r5、lenti-f1/r6、lenti-f1/r7、lenti-f2/r1、lenti-f1/r2、lenti-f1/r3、lenti-f1/r4、lenti-f1/r5、lenti-f1/r6、lenti-f1/r7、lenti-f3/r1、lenti-f3/r2、lenti-f3/r3、lenti-f3/r6、lenti-f3/r7)测试扩增特异性,测试后有6对引物可扩增含有sgrna序列的目标区域,其引物对序列如下:(1)lenti-f1:5’-cttgtggaaaggacgaaacacc-3’(seqidno.:132),lenti-r1:5’-caagttgataacggactagcc-3’(seqidno.:135);(2)lenti-f1:5’-cttgtggaaaggacgaaacacc-3’(seqidno.:132),lenti-r6:5’-aagcaccgactcggtgcca-3’(seqidno.:140);(3)lenti-f2:5’-atatcttgtggaaaggacg-3’(seqidno.:133),lenti-r1:5’-caagttgataacggactagcc-3’(seqidno.:135);(4)lenti-f2:5’-atatcttgtggaaaggacg-3’(seqidno.:133),lenti-r6:5’-aagcaccgactcggtgcca-3’(seqidno.:140);(5)lenti-f3:5’-gaaagtatttcgatttcttggc-3’(seqidno.:134),lenti-r1:5’-caagttgataacggactagcc-3’(seqidno.:135);(6)lenti-f3:5’-gaaagtatttcgatttcttggc-3’(seqidno.:134),lenti-r6:5’-aagcaccgactcggtgcca-3’(seqidno.:140).pcr反应体系为:5xprimestargxlbuffer:2μl;dntp(mg2+plus):0.8μl;forwardprimer:0.2μl;reverseprimer:0.2μl;dna:(20~50)ng;补水至10μl。反应程序为:94℃,5min;28个循环(98℃,10s;62℃,15s;68℃,10s);68℃,5min;15℃,2min。pcr检测结果如图12所示。6对引物可扩增含有sgrna序列的目标区域,其中3对引物对可进行特异性扩增,且扩增序列通过测序鉴定为插入的目的片段,基于此,为了达到最佳检测效果,优选引物对组合为:lenti-f1/r6或lenti-f3/r6。最佳为lenti-f1/r6。将pcr产物送公司构建含有sgrna序列区域文库和进行高通量测序,获得测序数据后进行生物信息学分析,最后确定质粒库中含有82392条sgrna,与上述设计的文库相比覆盖率为96.17%(82392/85674=96.17%),其中靶向14958个蛋白编码基因,14168个lincrna和142个mirna,并含有970个sgrna-nc最为阴性对照。由此可见,通过本发明方法,可获得了高覆盖度猪的全基因组crispr/cas9敲除文库。其中文库的部分序列见图16。实施例7构建稳定表达cas9的pk-15细胞系(pk15-cas9)及其活性鉴定采用慢病毒lenti-cas9-puromycin感染猪pk-15细胞,通过puro(3μg/ml)药物筛选获得稳定表达cas9的细胞株,通过转染靶向anpep基因的sgrna质粒(lenti-sgrna-gfp),sgrna序列为:gcaacagcgttgtgggtaggcgg(划线部分指sgrna所针对(对应)的靶序列结合位点所对应的pam)。转染48h后提取细胞dna,通过pcr扩增目的片段,引物对为:anpep-952-f:5’-ctcctgcagcctgtaaccaga-3’(seqidno.:142);anpep-952-r:5’-aaggttccaaggtcccgaatc-3’(seqidno.:143),发现不同克隆均有较高活性(图13),随机选择其中14#克隆株作为后续实验。实施例8慢病毒载体表达sgrna活性测试利用靶向anpep基因的慢病毒sgrna感染pk15-cas9细胞系,分别在感染后的2d,4d,6d,8d,10d,检测gfp表达和sgrna活性变化。其中,sgrna活性通过常规的t7e1酶切实验进行检测,并以切割活性百分比(indel%)表示。结果如图14(a,b)所示。感染慢病毒sgrna可引导cas9靶向切割,随着感染时间增加,gfp荧光逐渐增强,且在6dpi时,sgrna活性趋于稳定(约30%)。实施例9慢病毒sgrna文库活性测试将慢病毒sgrna文库(moi=0.3)感染pk15-cas9细胞系,然后接种单克隆细胞。生长至7-10d时,挑取表达gfp的单克隆细胞克隆团至12孔细胞培养板中。待细胞长满时提取细胞基因组dna,用引物对lenti-f1:atatcttgtggaaaggacg,lenti-r4:caagttgataacggactagcc进行扩增,目的是扩增整合到基因组的sgrna序列,扩增酶选用primestargxlpolymerase(takara,r050a),退火温度为60℃,延伸时间为15s。然后纯化pcr产物,克隆至pmd19-t载体,挑取菌落进行测序,以鉴定稳定整合进入细胞的sgrna。再根据sgrna序列确定其对应的基因,设计引物扩增基因sgrna靶序列及侧翼序列,用t7e1实验检测基因组编辑效率。测序结果如下:单克隆colony1:>enssscg00000021964_16_50164110_50164139_pcagcttgatgagaagaggc(seqidno.:156);单克隆colony2:>enssscg00000005129_1_221066930_221066959_mgttcgaccagggacttctga(seqidno.:157);单克隆colony3:>enssscg00000005037_1_202467280_202467309_pgcttgcagatttaattccat(seqidno.:158);单克隆colony4::>ensssct00000021494_62_91_mgatttggtgagcaagctacg(seqidno.:159);单克隆colony5:>enssscg00000003717_6_103771733_103771762_mttcatcttgaagcttggca(seqidno.:160);单克隆colony6:>enssscg00000013016_2_6384602_6384631_patccacgtatctcttggcc(seqidno.:161);单克隆colony7:>anpep-sgrlib-2accctacctcactcccaacg(seqidno.:162)。根据确定的基因设计扩增引物,如下:(1)colony1-1003-ftgtttccctccctggcaattt(seqidno.:144)colony1-1003-rttgccgctgtctcggtagaa(seqidno.:145)(2)colony2-1166-faggagaggtgcgggaatgt(seqidno.:146)colony2-1166-rtacttgctttgggaaatacaggc(seqidno.:147)(3)colony3-722-fggtgcaaatatcaagccagca(seqidno.:148)colony3-722-rcagactgtggcaattgactgt(seqidno.:149)(4)colony4-743-facctgtgactatccttcagcac(seqidno.:150)colony4-743-raacagctaacctccaggcac(seqidno.:151)(5)colony5-864-fggagtggttcttcttgctcca(seqidno.:152)colony5-864-ratcctggtttacagagttgtga(seqidno.:153)(6)colony6-1110-fagtctcagctaccaggccata(seqidno.:154)colony6-1110-rtgttcaggcagagatcacca(seqidno.:155)(7)colony7-952-fctcctgcagcctgtaaccaga(seqidno.:142)colony7-952-raaggttccaaggtcccgaatc(seqidno.:143)pcr扩增目的片段后,利用pcr产物纯化回收试剂盒,按照操作说明回收,用t7e1实验酶切,见图15,可见有5个细胞克隆株靶标位点被切割,且其中有两个细胞克隆株中目标靶点有大片段序列缺失。研究结果表明,本发明中猪的sgrna文库可用于构建突变体细胞库,能够用于全基因组水平功能基因筛选。在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1