一种高产率1,3-二苯基丙二酮类化合物的生产工艺的制作方法
本发明涉及精细化工领域,特别涉及一种高产率1,3-二苯基丙二酮类化合物的生产工艺。
背景技术:
近年来,紫外线辐射量正随着臭氧层的破坏而不断增加,其中320~400nm(简称uv-a)的近紫外光对人体会造成辐射伤害,诱发基因突变产生皮肤癌。人们为保护人体免受过量的紫外线辐射,把具有安全、高效吸收紫外线辐射的紫外线吸收剂广泛应用于化妆品、护发品、织物处理及洗涤剂中。紫外线吸收剂中包括有已知的1,3-二苯基丙二酮类化合物,如丁基甲氧基二苯甲酰甲烷(parsol1789,阿伏苯宗),其是专门用于护肤化妆品的一类抗uv-a紫外光线吸收剂,parsol1789最初由罗氏公司开发生产,产品命名为:parsol1789。该产品主要用于日用化妆品防晒霜、防晒剂等防晒系列产品中,还可作为一种稳定剂防止化妆品和香水的光分解作用。parsol1789在化妆品中的作用就是防止日光中紫外线uv-a对皮肤的辐射。1,3-二苯基丙二酮类化合物的紫外线吸收作用是依靠分子结构中酮式与烯醇式结构的转换,从而把吸收的光能转换成热能。其酮式异构体的λmax约为260nm,烯醇式异构体的λmax约为350nm正好位于uv-a波段,在两者互变异构的过程中可以吸收高能紫外线uv-a。1,3-二苯基丙二酮类化合物中的parsol1789是世界上仅有的少量几种uv-a型吸收剂中最为有效的一种,被美国fda列为非处方药(otc),准许在美国使用,欧盟和日本也已经批准其使用,为安全有效的防晒剂。因此,实现1,3-二苯基丙二酮类化合物,尤其是巴松1789工业化生产,对提高经济效益,促进企业发展有重要的意义。
经调研文献发现,1,3-二苯基丙二酮类化合物如防晒剂阿伏苯宗的合成方法有以下几种:
(1)以对甲氧基苯乙酮和对叔丁基苯甲酸甲酯为原料的合成方法是以强碱做催化剂进行直接缩合(日用化学工业,2009,39(3):179-182;精细石油化工,2009,26(3):4-7;化学世界,2006,28(9):538-540;cn1958549;香料香精化妆品,2002,3:8-10),用此法生产大体都用氨基钠、氢化钠或钠做催化剂。但是氨基钠在反应过程中易产生氨解等副反应,产品产率低;氢化钠和钠遇水剧烈反应,生产过程中安全隐患很大,对设备的要求也较高;且氨基钠和氢化钠价格昂贵,大规模生产需要的成本较高,在市场中无价格优势。
(2)以对叔丁基苯甲醛与对甲氧基苯乙酮在甲醇钠作用下缩合生成3-[4-叔丁基苯基]-1-(4-甲氧基苯基)-2-丙烯-1-酮;再经卤素加成,甲醇钠脱卤素合成(合成化学,2008,16(3):342-343;eur.pat.appl.994092),此法产率较高,但此法合成中需要用到溴素或者氯气,不仅对设备要求高,而且会对人和环境造成严重伤害。
(3)以对叔丁基苯乙酮和对甲氧基苯酰氯为原料,进行酰基化反应生成阿伏苯宗(wo2006100225),用此法合成中,需用cucl做催化剂,反应时间达20h,后处理繁琐,还需用到氯仿等毒性较大的试剂。
上述的方法都各有优缺点,因此,仍需继续探索新的合成方法,以适应业界对高品质、低价格的1,3-二苯基丙二酮类化合物的需求。
技术实现要素:
为了解决上述问题,本发明人进行了锐意研究,结果发现:反应过程中采用合适的催化剂和投料方式,并不断蒸出非目标产物,即可保证高纯度、高产率产品,同时简化生产工艺,后处理简单,对设备要求低,从而完成了本发明。
本发明的目的在于提供以下方面:
(1)一种1,3-二苯基丙二酮类化合物的生产工艺,该生产工艺包括以下步骤:
步骤1,将苯甲酸酯化合物a、催化剂和溶剂i加入反应釜中;
步骤2,向步骤1中体系中加入苯乙酮类化合物b,进行反应;
步骤3,反应结束后进行后处理,得到最终产物。
其中,所述苯甲酸酯化合物a的结构为:
r2为氢、烷基或烷氧基,所述烷基选自-ch3、-ch2ch3、-ch2(ch2)2ch3、-ch(ch3)2或-c(ch3)3,所述烷氧基选自-och3、-och2ch3或-och(ch3)2。
所述苯乙酮类化合物b的结构为:
根据本发明提供的一种1,3-二苯基丙二酮类化合物的生产工艺,具有以下有益效果:
(1)本发明采用甲醇钠作为主催化剂,使用少量的甲醇钾作为助催化剂,可明显提高产品1,3-二苯基丙二酮类化合物的收率;
(2)采用精馏柱,将反应副产物分馏出反应体系,不仅保证了反应体系的温度,而且改变了反应的平衡,使得反应向生成产品1,3-二苯基丙二酮类化合物的方向移动,从而缩短了反应时间,减少了副反应和杂质的生成;
(3)使用混合碱性催化剂,和带精馏柱反应,比单独使用甲醇钠和传统的反应方式收率要高出10%~15%,收率≥85.0%,从而使得副产物和三废大大减少;后处理经过酸化、洗涤、脱溶、结晶,最终产品颜色浅、质量好、市场竞争力强,易于工业化推广。
附图说明
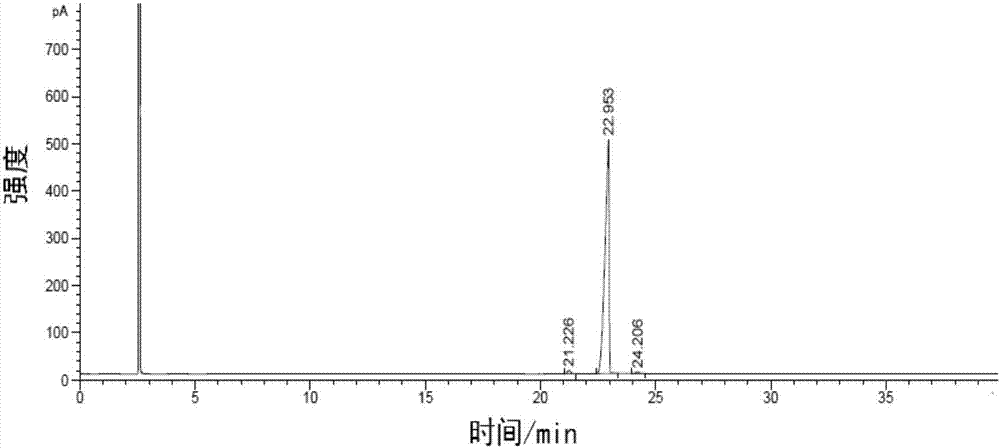
图1为实施例1中制备得到的阿伏苯宗的气相色谱图;
图2为对比例1中制备得到的阿伏苯宗的气相色谱图;
图3为对比例2中制备得到的阿伏苯宗的气相色谱图;
图4为对比例3中制备得到的阿伏苯宗的气相色谱图。
具体实施方式
下面通过对本发明进行详细说明,本发明的特点和优点将随着这些说明而变得更为清楚、明确。
在这里专用的词“示例性”意为“用作例子、实施例或说明性”。这里作为“示例性”所说明的任何实施例不必解释为优于或好于其它实施例。尽管在附图中示出了实施例的各种方面,但是除非特别指出,不必按比例绘制附图。
本发明的目的是提供一种1,3-二苯基丙二酮类化合物的生产工艺,该生产工艺包括以下步骤:
步骤1,将苯甲酸酯化合物a、催化剂和溶剂i加入反应釜中;
步骤2,向步骤1中体系中加入苯乙酮类化合物b,进行反应;
步骤3,反应结束后进行后处理,得到最终产物。
步骤1中,将苯甲酸酯化合物a、催化剂和溶剂i加入反应釜中,升温至设定温度。
其中,所述苯甲酸酯化合物a的结构为:
r2为氢、烷基或烷氧基,所述烷基选自-ch3、-ch2ch3、-ch2(ch2)2ch3、-ch(ch3)2或-c(ch3)3,所述烷氧基选自-och3、-och2ch3或-och(ch3)2。
在一种优选的实施方式中,所述催化剂为碱性催化剂,所述碱性催化剂为固体碱性催化剂或液体碱性催化剂。
碱性催化剂种类较多,选择的碱性催化剂不仅考虑其催化性能,同时还需考虑其在反应过程中是否产生分解、与反应物或目标产物之间的反应、生产过程中是否存在安全隐患、对设备是否有特殊要求、使用成本是否符合大规模生产要求等多种因素,鉴于对上述因素的考量,本发明人进行了大量的实验研究,确定了符合本发明的碱性催化剂。
在一种优选的实施方式中,所述催化剂选自甲醇钠或甲醇钾中任意一种或其组合;优选地,所述催化剂为固体甲醇钠与固体甲醇钾、固体甲醇钠与液体甲醇钾、液体甲醇钠与固体甲醇钾、或者液体甲醇钠与液体甲醇钾组成的混合碱性催化剂;更优选地,所述催化剂为固体甲醇钠与液体甲醇钾组成的混合催化剂。
在进一步优选的实施方式中,液体碱性催化剂的浓度为10~32(重量)%,优选浓度为28~32(重量)%;相应的,液体甲醇钾的甲醇溶液的浓度为10~32(重量)%,优选浓度为28~32(重量)%。
在更进一步优选的实施方式中,液体甲醇钾中甲醇钾与固体甲醇钠的重量比为(0.01~0.1):1,优选为(0.06~0.09):1。甲醇钾的碱性高于甲醇钠,且甲醇钾的成本也远高于甲醇钠,两者的重量比在此范围内,碱性满足反应要求,不会由于碱性过弱使得反应速率低反应时间长;也不会由于碱性过强使反应难以控制,同时带来成本的提升。
由上述可知,本发明中催化剂优选为混合碱性催化剂。现有技术中尚未有采用混合碱性催化剂制备1,3-二苯基丙二酮类化合物的工艺。在生产中,混合碱性催化剂的加入方式普遍为同时加入反应体系中,在同时段共同催化反应,这种方式操作方便,初始催化性能强,反应速度快,但持续性可能不足;为保证后期持续性,催化剂往往在初始加入较大量,这又容易导致副反应产生。
在一种优选的实施方式中,所述混合碱性催化剂分阶段加入。步骤1中将苯甲酸酯化合物a和甲醇钠优选固体甲醇钠加入反应釜中;通过甲醇钠催化苯甲酸酯化合物a与苯乙酮类化合物b反应。此时甲醇钠起到诱发反应的目的,固体甲醇钠相对于液体甲醇钠在体系中的浓度更高,在反应初始阶段更易促进反应进行;而且甲醇钠碱性较甲醇钾弱,不易引发原料自缩合反应等副反应;除此之外,由于碱性甲醇钠的存在,产物1,3-二苯基丙二酮类化合物在体系中以不溶于反应液的钠盐形式存在,体系为固液两相,促进了反应的进行。初始加入的甲醇钠为甲醇钠总重量的1/2~2/3。
在进一步优选的实施方式中,在甲醇钠单独催化设定时间后(或根据苯乙酮类化合物b反应程度确定)向反应体系中加入甲醇钾优选液体甲醇钾。液体甲醇钾碱性强,其加入可有效促进反应进行,减少反应时间。此时,甲醇钾优选液体甲醇钾可同时全部加入反应体系中或逐量慢慢加入,优选逐量加入甲醇钾。
在更进一步优选的实施方式中,在反应后期,再次加入甲醇钠优选甲醇钠固体,保证后续催化反应速率。
上述催化剂的加入方式可有效保证整个反应高效的催化效率,同时降低催化剂大量加入引发副反应的可能,使得最终产物纯度好、产率高。
在一种优选的实施方式中,加入的全部催化剂与苯甲酸酯化合物a的摩尔比为(0.35~3):1,优选为(1.10~1.35):1。在上述范围内,催化剂可有效实施催化效果,若摩尔比低于0.35:1,催化速率低;若摩尔比高于3:1,催化剂过量,会使反应体系进一步变得粘稠,使传质传热变差,影响产品的收率;同时增加了催化剂的使用成本,催化效果提升不明显。
在一种优选的实施方式中,所述溶剂i为醇类溶剂或烃类溶剂,优选为烃类溶剂,更优选为芳香烃类溶剂。
在进一步优选的实施方式中,所述的芳香烃类溶剂为苯、甲苯、乙苯、邻二甲苯、间二甲苯或对二甲苯,优选为甲苯。
在更进一步优选的实施方式中,所述溶剂i与苯甲酸酯化合物a的重量比为(2~10):1,优选(5~7):1。
在一种优选的实施方式中,将苯甲酸酯化合物a、催化剂和溶剂i加入反应釜中后,升温至90~115℃。
步骤1中反应釜温度即为后续反应温度,反应温度对于控制反应速率和副产物的生成有重要影响;反应温度升高,反应速率加快;反应温度高于115℃,则苯甲酸酯化合物a或后续加入的苯乙酮类化合物b易产生自缩合反应,降低最终产物的产率和纯度。
在进一步优选的实施方式中,将苯甲酸酯化合物a、催化剂和溶剂i加入反应釜中后,升温至100~110℃。
在更进一步优选的实施方式中,将苯甲酸酯化合物a、催化剂和溶剂i加入反应釜中后,升温至105~108℃。
在一种优选的实施方式中,所述反应釜为带精馏柱的反应釜。所述精馏柱为玻璃或不锈钢制成,其内堆砌填料,使得精馏柱的理论塔板数n≥20。精馏柱可不断分离出液体催化剂中的溶剂和反应过程中产生的低沸点副产物,使反应平衡向生成最终产物的方向进行,且副产物的分离简化了体系的组成,便于控制反应釜温度稳定维持在反应温度范围内。
同时,由于步骤2中加入的原料苯乙酮类化合物b的沸点略高于反应温度,采用带精馏柱的反应釜,在蒸出副产物时,不会伴随原料苯乙酮类化合物b的蒸出。
在进一步优选的实施方式中,所述填料为θ环不锈钢填料。θ环不锈钢填料由100目不锈钢丝网卷至成型,由于不锈钢丝网的毛细作用,液体能很好的分散成膜,利于气液两相进行充分传质、传热,可显著消除沟流等不稳定现象。
在更进一步优选的实施方式中,精馏柱的理论塔板数n为40≥n≥20。理论塔板数<20,分离时间短但分离效果差;而理论塔板数>40,分离效果满足要求,但时间长,效率低。
步骤2中,向步骤1中体系中加入苯乙酮类化合物b,进行反应。
其中,所述苯乙酮类化合物b的结构为:
-ch2(ch2)2ch3、-ch(ch3)2或-c(ch3)3,所述烷氧基选自-och3、-och2ch3、-och2ch2ch3或-och(ch3)2。
本发明的反应方程式为:
在一种优选的实施方式中,所述苯乙酮类化合物b溶于溶剂ii,以液态形式滴加加入反应釜中。
在一种优选的实施方式中,所述溶剂ii为醇类溶剂或烃类溶剂,优选为烃类溶剂,更优选为芳香烃类溶剂,最优选与溶剂i为相同溶剂。
溶剂i和溶剂ii的用量直接关系到反应速率和最终产物的产率,经过研究发现,溶剂ii与对苯乙酮类化合物b的重量比为(1~10):1时,反应速率较快,最终产物的产率较高。
在进一步优选的实施方式中,溶剂ii与对苯乙酮类化合物b的重量比为(1~3):1。
在一种优选的实施方式中,所述苯乙酮类化合物b与所述苯甲酸酯化合物a的重量比为(0.7~0.9):1。为使反应充分进行,选择苯甲酸酯化合物a过量。同时,由于体系不是绝对无水,因此,甲醇钠加入会有很少量的氢氧化钠产生;此外,甲醇钠本身也含有游离碱,如氢氧化钠,碳酸钠等,因此,反应体系始终有游离碱存在,苯甲酸酯化合物a发生皂化反应不可避免,使得苯甲酸酯化合物a的投料量需过量。
在进一步优选的实施方式中,所述苯乙酮类化合物b与所述苯甲酸酯化合物a的重量比为(0.75~0.78):1。
在一种优选的实施方式中,所述苯乙酮类化合物b的滴加时间为1~3h。值得注意的是,苯乙酮类化合物b的滴加时间需严格控制在上述范围内,若滴加时间<1h,苯乙酮类化合物b的局部浓度高,在反应釜中会发生自缩合反应;若滴加时间>3h,加之后续的保温反应时间,产物在反应温度下易发生分解;上述两种情况,不论反应原料的自缩合或者反应最终产物的分解,均会造成产率下降,以及杂质去除难度加大的问题。
在进一步优选的实施方式中,所述苯乙酮类化合物b的滴加时间为1~2.5h。
在更进一步优选的实施方式中,所述苯乙酮类化合物b的滴加时间为1~1.5h。
在更进一步优选的实施方式中,苯乙酮类化合物b的滴加过程中,通过精馏柱不断分馏出副产物r1oh。
在一种优选的实施方式中,滴加结束后,在90~115℃下继续保温反应1~2h,保证反应原料充分反应。
在进一步优选的实施方式中,保温过程中通过精馏柱不断分馏出副产物r1oh。
步骤3中,反应结束后进行后处理,得到最终产物。
在一种优选的实施方式中,所述后处理包括酸化、水洗、溶剂去除和重结晶。
最终产物中两酮基间的亚甲基活泼,易于过量的催化剂发生反应,使最终产物在反应体系中以钾盐或钠盐的形式存在。因而,反应完成需加入酸,以得到非盐形式的最终产物。所述酸为无机酸,选自稀盐酸(10重量%~12重量%)、稀硫酸(10重量%~12重量%)、稀硝酸(10重量%~12重量%)或醋酸。
在一种优选的实施方式中,酸化过程中伴随搅拌,加快反应进行。
在一种优选的实施方式中,酸化后,分去水层,有机相水洗多次,除去其中残留的酸。
在一种优选的实施方式中,采用真空蒸馏脱除有机相中的溶剂i和溶剂ii,得到粗产品。
向粗产品中加入结晶溶剂,所述结晶溶剂为醇类溶剂或烃类溶剂。
在进一步优选的实施方式中,所述的醇类溶剂为甲醇、乙醇或异丙醇中任意一种或其组合,优选为甲醇。
在更进一步优选的实施方式中,所述结晶溶剂与苯甲酸酯化合物a的重量比为(3~6):1。在上述用量范围内的结晶溶剂,可有效去除最终产物中的杂质,提高最终产物的纯度,且使得最终产物颜色合格,具有特征性的芳香气味;不会由于溶剂过少导致最终产物质量低,或者溶剂用量多,影响结晶量,增加溶剂消耗。
在更进一步优选的实施方式中,结晶溶剂与苯甲酸酯化合物a的重量比为(3.5~4):1。
实施例
以下以阿伏苯宗的生产工艺为例,通过具体实例进一步描述本发明。不过这些实例仅仅是范例性的,并不对本发明的保护范围构成任何限制。
实施例1
在带精馏柱(堆砌θ环不锈钢填料,理论塔板数20)的反应釜中加入800g甲苯(toluene)、24g甲醇钠、6g32%的液体甲醇钾溶液和80g对叔丁基苯甲酸甲酯,边升温边分馏出甲醇,至釜温达到105~108℃;
开始滴加60g对甲氧基苯乙酮和80g甲苯组成的混合溶液,并同时从精馏柱塔顶分馏出反应副产的甲醇,以维持釜温在105~108℃,滴加时间控制在1.5h。滴加完成后,保温反应2h,保温过程中,不断从精馏柱塔顶分馏出反应副产的甲醇,以维持釜温在105~108℃。
反应结束后,加入10(重量)%的稀硫酸水溶液120g,搅拌洗涤后分去水层,有机层用300g水分三次洗涤。蒸馏脱去溶剂甲苯,馏底加入300g甲醇析晶,过滤,滤饼重结晶得类白色针状晶体阿伏苯宗。反应方程式如下:
上述方法制备得到的阿伏苯宗gc纯度(面积归一法)为98.2398%,重量含量为99.2%,收率为85.6%,气相色谱图如图1所示。
实施例2
在带精馏柱(堆砌θ环不锈钢填料,理论塔板数20)的反应釜中加入800g甲苯、12g甲醇钠、80g对叔丁基苯甲酸甲酯,边升温边分馏出甲醇,至釜温达到105~108℃;
开始滴加60g对甲氧基苯乙酮和80g甲苯组成的混合溶液,并同时从精馏柱塔顶分馏出反应副产的甲醇,以维持釜温在105~108℃,滴加时间控制在1.5h,滴加0.5h后加入6g32%的液体甲醇钾溶液。滴加完成后,再次加入12g甲醇钠,保温反应2h,保温过程中,不断从精馏柱塔顶分馏出反应副产的甲醇,以维持釜温在105~108℃。
反应结束后,加入10(重量)%的稀硫酸水溶液120g,搅拌洗涤后分去水层,有机层用300g水分三次洗涤。蒸馏脱去溶剂甲苯,馏底加入300g甲醇析晶,过滤,滤饼重结晶得类白色针状晶体阿伏苯宗。
上述方法制备得到的阿伏苯宗gc纯度(面积归一法)为98.7461%,重量含量为99.4%,收率为92.8%。
对比例
对比例1
与实施例1比较,不采用精馏柱且不蒸馏出反应副产的甲醇。
在不带精馏柱的反应釜中加入800g甲苯、24g甲醇钠、6g32%的液体甲醇钾溶液和80g对叔丁基苯甲酸甲酯,升温至回流(98~103℃);
开始滴加60g对甲氧基苯乙酮和80g甲苯组成的混合溶液,滴加过程中始终保持回流,釜温逐渐下降,滴加时间控制在1.5h。滴加完成后,保温(80~83℃)反应2h。
反应结束后,加入10(重量)%的稀硫酸水溶液120g,搅拌洗涤后分去水层,有机层用300g水分三次洗涤。蒸馏脱去溶剂甲苯,馏底加入300g甲醇析晶,过滤,滤饼重结晶得类白色针状晶体阿伏苯宗
上述方法制备得到的阿伏苯宗gc纯度(面积归一法)为98.2244%,重量含量为98.7%,收率为65.6%,气相色谱图如图2所示。
对比例2
与实施例1比较,不采用精馏柱,但反应过程中不断蒸出反应副产的甲醇。
在不带精馏柱的反应釜中加入800g甲苯、24g甲醇钠、6g32%的液体甲醇钾溶液和80g对叔丁基苯甲酸甲酯,边升温边分馏出甲醇,至釜温达到105~108℃;
开始滴加60g对甲氧基苯乙酮和80g甲苯组成的混合溶液,并同时蒸馏出反应副产的甲醇,以维持釜温在105~108℃,滴加时间控制在1.5h。滴加完成后,保温反应2h,保温过程中,不断蒸馏出反应副产的甲醇,以维持釜温在105~108℃。
反应结束后,加入10(重量)%的稀硫酸水溶液120g,搅拌洗涤后分去水层,有机层用300g水分三次洗涤。蒸馏脱去溶剂甲苯,馏底加入300g甲醇析晶,过滤,滤饼重结晶得类白色针状晶体阿伏苯宗。
上述方法制备得到的阿伏苯宗gc纯度(面积归一法)为98.2327%,含量为99.4%,收率为73.7%,气相色谱图如图3所示。
对比例3
与实施例1比较,不采用精馏柱,但反应过程中不断蒸出反应副产的甲醇;不加入甲醇钾而以单一的甲醇钠做催化剂。
在不带精馏柱的反应釜中加入800g甲苯、25.5g甲醇钠、和80g对叔丁基苯甲酸甲酯,边升温边分馏出甲醇,至釜温达到105~108℃;
开始滴加60g对甲氧基苯乙酮和80g甲苯组成的混合溶液,并同时蒸馏出反应副产的甲醇,以维持釜温在105~108℃,滴加时间控制在1.5h。滴加完成后,保温反应2h,保温过程中,不断蒸馏出反应副产的甲醇,以维持釜温在105~108℃。
反应结束后,加入10(重量)%的稀硫酸水溶液120g,搅拌洗涤后分去水层,有机层用300g水分三次洗涤。蒸馏脱去溶剂甲苯,馏底加入300g甲醇析晶,过滤,滤饼重结晶得类白色针状晶体阿伏苯宗。
上述方法制备得到的阿伏苯宗gc纯度(面积归一法)为98.2013%,含量为99.3%,收率为80.1%,气相色谱图如图4所示。
以上结合具体实施方式和范例性实例对本发明进行了详细说明,不过这些说明并不能理解为对本发明的限制。本领域技术人员理解,在不偏离本发明精神和范围的情况下,可以对本发明技术方案及其实施方式进行多种等价替换、修饰或改进,这些均落入本发明的范围内。本发明的保护范围以所附权利要求为准。
- 还没有人留言评论。精彩留言会获得点赞!