一种盐酸沙丙蝶呤的制备方法与流程
本发明涉及一种盐酸沙丙蝶呤(sapropterindihydrochloride)的制备方法,属于药物合成领域。
背景技术:
苯丙酮尿症(phenylketonuria,pku)是一种常染色体隐性遗传性疾病.因肝脏苯丙氨酸羟化酶缺乏或者四氢生物蝶呤合成酶、二氢生物喋呤还原酶缺陷而导致苯丙氨酸代谢障碍。目前盐酸沙丙蝶呤是pku的唯一治疗药物,它是人工合成的四氢生物蝶呤(bh4),是苯丙氨酸、酪氨酸、色氨酸三种芳香族氨基酸羟化酶的辅酶,使用盐酸沙丙蝶呤治疗pku被称为bh4替代疗法。
盐酸沙丙蝶呤,化学名为(6r)-2-氨基-6-[(1r,2s)-1,2-二羟丙基]-5,6,7,8-四氢-4(1h)-蝶啶二盐酸盐,结构式如下式(i):
(i)
目前,盐酸沙丙蝶呤的常用路线(helveticachimicaacta,1985,68,1639-1643和us2015291590a1)如下:
该路线以5-脱氧-l-阿拉伯糖(dl-sma)为原料,先与苯肼缩合制得苯腙类化合物(dl-6),然后其经过乙酰化得到2,3,4-o-三乙酰基-5-脱氧-l-阿拉伯糖苯腙(dl-2),然后苯腙与2,4,5-三氨基-6-羟基嘧啶硫酸盐(dl-smb)环合、氧化、水解、氢化得到终产品盐酸沙丙蝶呤。该路线的缺点是dl-6化合物不稳定,在下一步上保护基时苯胺基容易脱去,生成较多杂质。根据us2015291590a1所述路线制得的终产品盐酸沙丙蝶呤的hplc图见说明书附图3,可以看到,终产品中有一个较大的杂质难以去除。并且该路线在处理中间体dl-3和dl-4时,都进行了柱层析,不适用于工业化生产。
专利cn101959891在合成中间体dl-5时,利用双相混合液水解,杂质留在有机相正丁醇中,水相含产物,纯度虽高收率较低,并且使用了令人反胃且易燃的有机溶剂正丁醇。
专利wo2016189542a1中,在合成中间体dl-5时,利用化合物能够成钾盐的性质,通过常规的调节水溶液ph值的方法,对中间体dl-5进行纯化,操作复杂,收率较低。
因此现有路线中均存在各种不足之处,我们需要一条更优化的路线来生产该药物。
技术实现要素:
针对现有技术存在的问题,本发明人经过大量的条件实验,修改工艺路线,优化中间体纯化精制方法,以dl-sma(5-脱氧阿拉伯糖)为原料,先乙酰化再在酸催化下与苯肼发生席夫碱化反应得dl-2,然后dl-2与氨基嘧啶(dl-smb)反应关环得中间态dl-3,通过一锅法氧化得到重要中间体dl-4,不经纯化在水溶液中直接水解得中间体dl-5,然后调节水溶液的ph值,最后进行离子树脂纯化,得到终产品。
具体地,本发明涉及一种式(i)表示的盐酸沙丙蝶呤的制备方法,具体为通过下式(ii)表示的反应步骤a-e得到盐酸沙丙蝶呤:
(i)
(ii)
a)以5-脱氧阿拉伯糖(dl-sma)为原料,经乙酰化反应得到中间体dl-1;
b)dl-1在酸催化下与苯肼反应,得到中间体dl-2;
c)dl-2与硫酸氨基密啶关环,得到中间体dl-3,不经纯化直接氧化后得到中间体dl-4;
d)dl-4经过水解得到l-生物喋呤(dl-5);
e)l-生物喋呤经氢化还原、成盐得到盐酸沙丙蝶呤。
优选的本发明的制备方法,其中步骤a的反应温度为0-20℃。
优选的本发明的制备方法,其中步骤b的反应液的初始ph值控制在3-7。
优选的本发明的制备方法,其中步骤c的关环工序的催化剂为选自高氯酸、高氯酸盐、和保险粉与醋酸盐的水溶液中的一种。
优选的本发明的制备方法,其中步骤c的氧化工序的氧化剂为碘单质或双氧水。
优选的本发明的制备方法,其中步骤d的水解工序使用强酸或强碱。
优选的本发明的制备方法,其中步骤d的纯化方法使用离子交换树脂。
优选的本发明的制备方法,其中步骤e中氢化的催化剂选自钯碳、铂黑、氧化铂中的一种。
优选的本发明的制备方法,其中步骤e中氢化的压力为2mpa-4mpa。
优选的本发明的制备方法,其中最为优选的制备方法为:5-脱氧阿拉伯糖(dl-sma)在乙酸酐存在下,经乙酰化反应得到中间体dl-1;dl-1在乙酸催化下与苯肼反应,得到中间体dl-2;dl-2与硫酸氨基密啶在无水高氯酸锂催化下关环,得到中间体dl-3,不经纯化直接被碘单质氧化后得到中间体dl-4;dl-4经氢氧化钾水解得到l-生物喋呤(dl-5);l-生物喋呤经铂催化剂还原、成盐得到盐酸沙丙蝶呤。
本发明的盐酸沙丙蝶呤的制备方法,具有如下优点:首先,步骤a和b中,5-脱氧阿拉伯糖先发生乙酰化再与苯肼反应,避免了生成不稳定的中间体dl-6,减少了下一步副反应的发生;其次,在中间体dl-4的合成过程中使用一锅法,减少了流程与工艺时间;再者,减少了对中间体dl-3和dl-4的精制,缩短了合成周期,减少了能耗,最后,在中间体dl-5的合成中,使用水作溶剂,避免了正丁醇的使用,对环境友好;在中间体dl-5的纯化过程中,通过离子交换树脂对其进行精制,所得产品纯度高,并且有益于提高终产品的纯度。整条路线操作简便,收率高,路线总收率70%以上,终产品纯度达99.5%以上,能耗低,适用于工业化生产。
附图说明:
图1为根据本发明的实施例5所制得的中间体dl-5的hplc图。
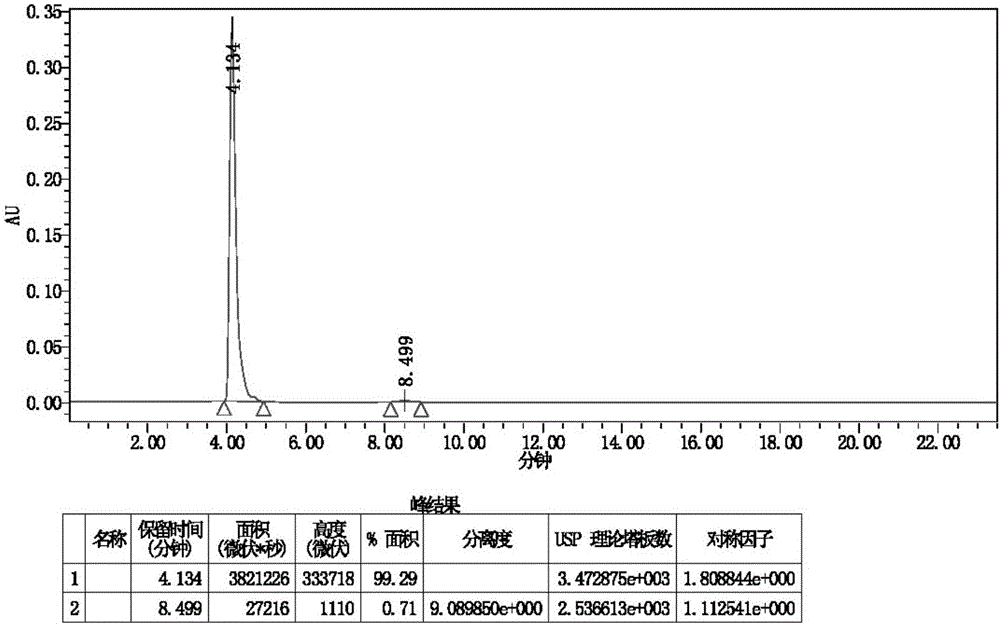
图2为根据本发明的实施例8所制得的终产品盐酸沙丙蝶呤的hplc图。
图3为根据专利us2015291590a1所述路线制得的终产品盐酸沙丙蝶呤的hplc图。
具体实施方式
以下结合具体实施例对本发明进行进一步的阐释。本发明的内容包括但不限于以下实施例。
实例1dl-1的合成:
向反应容器中依次加入dl-sma(134g,1.0mol),乙酸乙酯1.0l,4-二甲氨基吡啶24.4g,降温至10~15℃,滴加乙酸酐(326.7g,3.2mol),控制内温15~20℃,滴毕,控制反应温度0-20℃,tlc监控至反应完毕。后处理:使用饱和碳酸钠水溶液淬灭,分液后收集有机相,水相用乙酸乙酯萃取(500ml*2),合并有机相,再用饱和碳酸钠水溶液洗涤(400ml*1),收集有机相旋干,回收乙酸乙酯,得到淡黄色油状物dl-1(260g),直接进行下一步反应。
实例2dl-2的合成:
向反应容器中加入上步反应得到的dl-1(260g)和900ml水,滴加乙酸调节ph值在3-7,然后加入苯肼(118.8g,1.1mol,1.1eq),搅拌并在氮气保护下室温反应过夜。tlc监控,反应完毕后,反应液用乙酸乙酯萃取两次(500ml*2),合并有机相,无水硫酸钠干燥后滤去干燥剂,旋干得黄色油状物dl-2(340g),直接进行下一步反应。
实例3dl-4的合成方法1:
向反应容器中加入dl-2(181g,0.51mol),甲醇600ml,搅拌至全部溶解。加入无水高氯酸锂(1.8g,0.187mol),搅拌反应30分钟。控温30±5℃下,加入提前配好的dl-smb(151g,0.63mol)的氢氧化钠水溶液。氮气保护下,升温至回流,反应过夜,tlc监控至反应结束。降温至10±5℃,滴加溶有碘单质(328g,1.29mol)的甲醇(600ml)溶液,滴毕,室温下反应,tlc监控至反应完毕。过滤,用甲醇淋洗,然后水洗,所得固体50℃下干燥至恒重,得到浅棕色固体dl-4(300g)。
实例4dl-4的合成方法2:
向反应容器中加入dl-2(23g,66mmol),乙酸乙酯230ml,吡啶95.8ml搅拌至全溶。加入保险粉(1.7g,0.010mol)和乙酸钠(12.4g,0.151mol)的水溶液(100ml),搅拌30分钟。控温30±5℃,加入提前配好的dl-smb(18.4g,0.077mol)的水溶液(125ml)。氮气保护下,升温至回流,反应过夜,tlc监控至反应结束。降温至10±5℃,滴加双氧水溶液(30%,82g),滴毕,室温下反应,tlc监控至反应完毕。过滤,用甲醇淋洗,然后水洗,所得固体50℃下干燥至恒重,得到dl-4(20g)。
实例5dl-5的合成方法1:
向反应容器中加入氢氧化钾(164g,2.48mol),水(700ml),搅拌至全部溶解,降温至室温,加入dl-4(157.1g,0.486mol),搅拌至全部溶解,升温至40±5℃反应。tlc监控至反应完毕。停止加热,降温。滴加3n的hcl调节ph值至7-8,以离子交换树脂对其进行纯化,除去盐和其他杂质。旋蒸至干,得浅黄色固体(102g,0.43mol,收率88.5%)。
实例6dl-5的合成方法2:
向反应容器中加入水(700ml),浓盐酸(205ml),搅拌,室温下加入dl-4(157.1g,0.486mol),搅拌至全部溶解,升温至60℃。tlc监控至反应完毕。停止加热,降温。分液,收集水相,滴加3n氢氧化钠水溶液调节ph值至7-8,以离子交换树脂对其进行纯化,除去盐和其他杂质。旋蒸至干,得浅黄色固体(98g,41.3mol,收率85.2%)。
由实施例1-6可知,从dl-sma到dl-5,总收率约为85%以上。
实例7盐酸沙丙蝶呤的合成方法1:
向反应容器中加入dl-5(20g,0.084mol),水(200ml),磷酸氢二钾(7.2g),加入10m氢氧化钾水溶液调节ph值至12,dl-5基本全溶。将其转移至2l加氢釜中,加入无水钯碳(5g),氢气置换三次后,加氢至4mpa,室温反应24h,hplc监控至反应完毕。过滤除去催化剂,收集滤液,滴加浓盐酸,调节ph至1,旋蒸至剩余溶液约50ml,加入甲醇160ml,搅拌30min,过滤,收集滤液,旋蒸至剩余溶液约30ml,加入66ml浓盐酸,然后加入100ml乙醇,析出固体,过滤,用乙醇淋洗,收集固体,烘干,得白色晶体盐酸沙丙蝶呤17.5g,收率为86.4%,ee值为99%。
实例8盐酸沙丙蝶呤的合成方法2:
向反应容器中加入dl-5(20g,0.084mol),水(200ml),加入25%四乙基氢氧化铵水溶液,调节ph值至12,dl-5基本全溶。将其转移至2l加氢釜中,加入二氧化铂(2.0g),氢气置换三次后,加氢至2mpa,室温反应24h,hplc监控至反应完毕。过滤除去催化剂,收集滤液,加入浓盐酸,调节ph至1,旋干溶剂,加入无水乙醇(2000ml),搅拌1小时后过滤,收集滤液,旋干,得红棕色油状物22g。加入66ml浓hcl(10n),加热至全溶,滴加乙醇(220ml),析出固体,过滤,用乙醇淋洗,收集固体,烘干,得白色晶体盐酸沙丙蝶呤17.9g,收率为88.3%,ee值为99%。终产品盐酸沙丙蝶呤的hplc图见说明书附图2。
- 还没有人留言评论。精彩留言会获得点赞!