一种唑啉草酯关键中间体的制备方法与流程
本发明涉及生物制剂技术领域,特别涉及一种唑啉草酯关键中间体的制备方法。
背景技术
唑啉草酯(pinoxaden)是先正达开发的一种新苯基吡唑啉类除草剂,亦属于乙酰辅酶a羧化酶(acc)抑制剂类除草剂。唑啉草酯主要用于小麦及大麦田防除一年生禾本科杂草,对野燕麦、黑麦草、狗尾草、硬草、茼草、看麦娘、日本看麦娘、棒头草等禾本科杂草有良好防效,尤其对一些恶性禾本科杂草如野燕麦、黑麦草、硬草和茼草的防效接近100%。另外,它对小麦和大麦的安全性较高,施药适期宽(从麦苗2叶1心期至孕穗期均可施用),这些特点均使得唑啉草酯自上市以来获得了极快的发展。
唑啉草酯英文通用名为pinoxaden,商品名有:爱秀、axial等。其结构式如下,相对分子质量:400.5;分子式:c23h32n2o4;其iupac名为:8-(2,6-diethyl-p-tolyl)-1,2,4,5-tetrahydro-7-oxo-7h-pyrazolo[1,2-d][1,4,5]oxadiazepin-9-yl2,2-dimethylpropionate,即8-(2,6-二乙基-对甲苯基)-1,2,4,5-四氢-7-氧代-7h-吡唑并[1,2-d][1,4,5]氧杂二氮杂卓-9-基2,2-二甲基丙酸酯。
唑啉草酯的合成方法主要有以下几种:(1)通过有机锡试剂的stille偶联反应和氢化反应合成了主要中间体2-(2,6-二乙基-4-甲基苯基)丙二酸二甲酯;这里面首先一个问题就是催化剂昂贵的问题,而且均相催化剂[(ph3p)4]pd很难简单回收套用;其次有机锡试剂的毒性问题以及合成成本问题。(2)首先用2,6-二乙基-4-甲基苯胺重氮化再进行sandmeyer反应制备溴苯,继续与丙二腈在氯化钯/三苯基膦催化下偶联,最后用碳酸钾/双氧水得到酰胺。这里面同样一个问题也是催化剂昂贵的问题,均相催化剂[(ph3p)2]pdcl2很难简单回收套用;其次原料丙二腈的毒性问题以及合成成本问题。
上面合成方法中最关键的的就是中间体2-(2,6-二乙基-4-甲基苯基)丙二酸二甲酯或2-(2,6-二乙基-4-甲基苯基)丙二酰胺的合成。
因此,发明一种唑啉草酯关键中间体的制备方法来解决上述问题很有必要。
技术实现要素:
本发明的目的在于提供一种唑啉草酯关键中间体的制备方法,以解决上述背景技术中提出的问题。
为实现上述目的,本发明提供如下技术方案:一种唑啉草酯关键中间体的制备方法,其包括如下步骤:
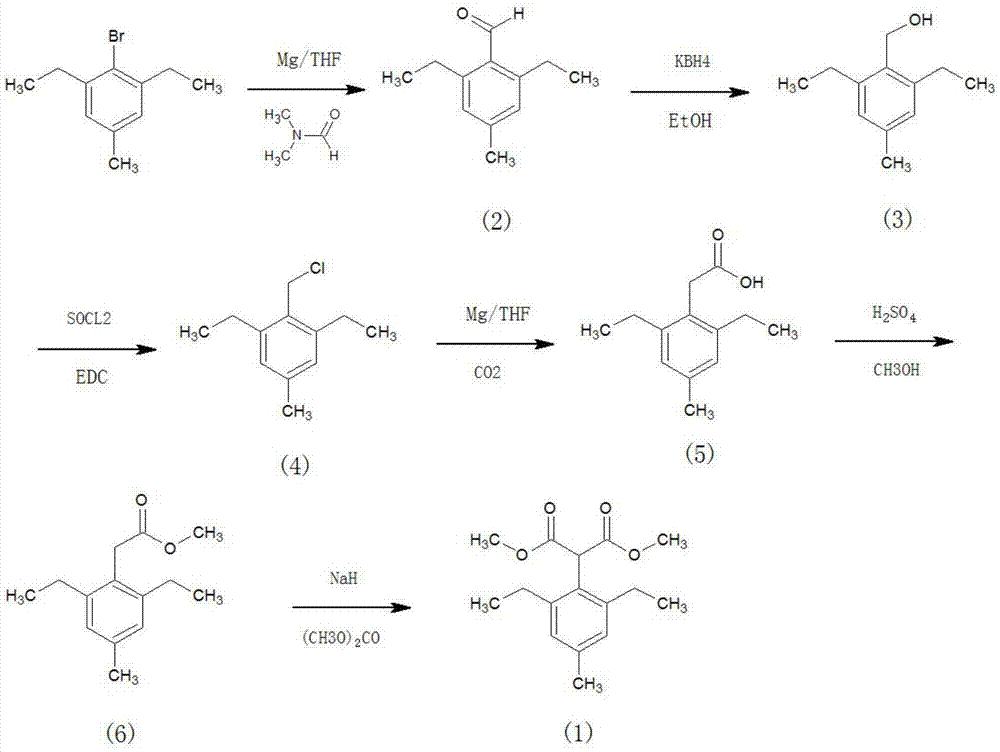
步骤一:利用2,6-二乙基-4-甲基溴苯为原料先与镁条在四氢呋喃溶剂中制备格式试剂再与n,n-二甲基甲酰胺反应,后处理加稀酸中和得到2,6-二乙基-4-甲基苯甲醛,再与硼氢化钾在乙醇中进行还原,还原产物用氯化亚砜作用得到2,6-二乙基-4-甲基苄基氯;
步骤二:用镁格式化通入二氧化碳得到2,6-二乙基-4-甲基苯乙酸,2,6-二乙基-4-甲基苯乙酸用甲醇酯化之后在碱作用下与碳酸甲酯反应得到2-(2,6-二乙基-4-甲基苯基)丙二酸二甲酯。
优选的,所述稀酸具体为10%的稀盐酸溶液。
优选的,所述步骤一中硼氢化钾还可用硼氢化钠替代。
本发明的技术效果和优点:
1、本发明设计了一条全新的该化合物合成路线,避免了贵重金属催化剂的使用引起的成本昂贵的问题;
2、在原料的选取和采用中杜绝了使用丙二腈高毒原料,提高了生产的安全性,有利于环保;
3、本发明中各步反应条件温和收率稳定,安全性能高,有利于进行工业化生产;
4、本发明工艺简单,设备要求低,可操作性强,具有良好的社会推广应用。
附图说明
图1为本发明合成工艺示意图。
图2为本发明的2,6-二乙基-4-甲基苯甲醛的合成工艺示意图。
图3为本发明的2,6-二乙基-4-甲基苄醇的合成工艺示意图。
图4为本发明的2,6-二乙基-4-甲基苄氯的合成工艺示意图。
图5为本发明的2,6-二乙基-4-甲基苯甲酸的合成工艺示意图。
图6为本发明的2,6-二乙基-4-甲基苯甲酸甲酯的合成工艺示意图。
图7为本发明的2,6-二乙基-4-甲基苯丙二甲酸二甲酯的合成工艺示意图。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1
如图1-7所示的一种唑啉草酯关键中间体的制备方法,其包括如下步骤:
步骤一:利用2,6-二乙基-4-甲基溴苯为原料先与镁条在四氢呋喃溶剂中制备格式试剂再与n,n-二甲基甲酰胺反应,后处理加稀酸中和得到2,6-二乙基-4-甲基苯甲醛,再与硼氢化钾在乙醇中进行还原,还原产物用氯化亚砜作用得到2,6-二乙基-4-甲基苄基氯;
步骤二:用镁格式化通入二氧化碳得到2,6-二乙基-4-甲基苯乙酸,2,6-二乙基-4-甲基苯乙酸用甲醇酯化之后在碱作用下与碳酸二甲酯反应得到2-(2,6-二乙基-4-甲基苯基)丙二酸二甲酯。
实施例2
与实施例1不同的是,本发明在进行具体生产中,部分原料可采取与其自身化学性质相类似的原材料进行更换和替代,其具体为:
用原甲酸三甲酯代替n,n-二甲基甲酰胺(dmf)反应含量84.7%。
利用2,6-二乙基-4-甲基溴苯为原料先与镁条在四氢呋喃溶剂中制备格式试剂再与原甲酸三甲酯反应,后处理加稀酸中和得到2,6-二乙基-4-甲基苯甲醛,再与硼氢化钾在乙醇中进行还原,还原产物用氯化亚砜作用得到2,6-二乙基-4-甲基苄基氯;
步骤二:用镁格式化通入二氧化碳得到2,6-二乙基-4-甲基苯乙酸,2,6-二乙基-4-甲基苯乙酸用甲醇酯化之后在碱作用下与碳酸甲酯反应得到2-(2,6-二乙基-4-甲基苯基)丙二酸二甲酯。
实施例3
与实施例1不同的是,本发明中,具体还包括以下步骤:
如图2所示,在500毫升三口瓶中,在氮气保护下加入四氢呋喃200ml,新活化的镁条12g,滴加1ml的2,6-二乙基-4-甲基溴苯升温到50℃引发反应,适当降温之后控制在30℃以下滴加100g的2,6-二乙基-4-甲基溴苯,滴完保温30分钟之后用冰水降温到0℃开始滴加100g的无水dmf,控制温度在10℃以下。滴完之后升到室温继续搅拌2小时,升温回收部份溶济之后倒入10%的稀盐酸溶液中并用乙酸乙酯萃取反应液,气相色谱分析含量95.3%。产品不需处理进入下一步反应。
如图3所示,在500毫升三口瓶中加入上述反应液,降温到0℃分批投入6g固体硼氢化钾,搅拌使反应完全tlc检测。
加水洗分层之后脱出溶济得到白色固体产品,75g,含量94.2%。
如图4所示,在500毫升三口瓶中加入300g二氯乙烷再投入36g的2,6-二乙基-4-甲基苄醇固体,降温到10℃滴加30g的氯化亚砜,滴加1-2小时,加完慢慢升温到回流,并保持回流3小时取样tlc检测反应完全。慢慢倒入500g的冰水中充分搅拌静止分层,有机层用10%的碳酸氢钠水溶液洗,再加200g水洗之后减压脱溶得到淡黄色油状液体40g,gc含量96.8%。
如图5所示,在500毫升三口瓶中,在氮气保护下加入四氢呋喃200ml,新活化的镁条5g,滴加1ml的2,6-二乙基-4-甲基苄氯到反应瓶中,在室温下搅拌30分钟直到引发反应,降温到-5℃之后开始滴加40g的2,6-二乙基-4-甲基苄氯并控制在0℃左右2-3小时滴加完,滴完保温30分钟之后自然升到室温再保温1小时,用冰水降温到0℃通入干燥的二氧化碳气体,控制温度在10℃以下,直到反应不再放热过量二氧化碳气体溢出停止通气。加入200g水并用10%的稀盐酸调节ph值到5之后过滤出白色固体产品,烘干得到38.6g气相色谱分析含量98.4%。
如图6所示,将20g的2,6-二乙基-4-甲基苯甲酸固体投入100g的无水甲醇与10g浓硫酸中加热回流6小时,减压回收过量的甲醇之后加入100g冰水中充分搅拌,加50g乙酸乙酯萃取二次再合并有机层,浓缩回收乙酸乙酯之后得到20g的2,6-二乙基-4-甲基苯甲酸甲酯含量96.1%。
如图7所示,在500ml反应并中加入100ml的无水碳酸二甲酯通氮气保护,分批加入nah(60%)10.9g。加完升温回流,同时滴加20g的2,6-二乙基-4-甲基苯甲酸甲酯与50g碳酸二甲酯的混合液,滴加约3小时,加完保持回流6小时之后,取样检测原料反应完全,用冰水降温到0℃以下,滴加5%的稀盐酸,调节ph值到1-2静止分层得到有机层,减压回收溶济之后加10g甲醇重结晶得以20g的白色固体产品,含量97.3%。
实施例4
与实施例3不同的是,本发明在进行具体生产中,部分原料可采取与其自身化学性质相类似的原材料进行更换和替代,其具体为:
用硼氢化钠(nabh4)代替第二步硼氢化钾得到同样收率含量;
2,6-二乙基-4-甲基苯甲酸甲酯的合成用对甲苯磺酸代替硫酸得到含量98.6%;
2,6-二乙基-4-甲基苯丙二甲酸二甲酯的合成用甲醇钠代替钠氢反应收率65%。
如图2所示,在500毫升三口瓶中,在氮气保护下加入四氢呋喃200ml,新活化的镁条12g,滴加1ml的2,6-二乙基-4-甲基溴苯升温到50℃引发反应,适当降温之后控制在30℃以下滴加100g的2,6-二乙基-4-甲基溴苯,滴完保温30分钟之后用冰水降温到0℃开始滴加100g的无水dmf,控制温度在10℃以下。滴完之后升到室温继续搅拌2小时,升温回收部份溶济之后倒入10%的稀盐酸溶液中并用乙酸乙酯萃取反应液,气相色谱分析含量95.3%。产品不需处理进入下一步反应。
如图3所示,在500毫升三口瓶中加入上述反应液,降温到0℃分批投入6g固体硼氢化钠,搅拌使反应完全tlc检测。
加水洗分层之后脱出溶济得到白色固体产品,75g,含量94.2%。
如图4所示,在500毫升三口瓶中加入300g二氯乙烷再投入36g的2,6-二乙基-4-甲基苄醇固体,降温到10℃滴加30g的氯化亚砜,滴加1-2小时,加完慢慢升温到回流,并保持回流3小时取样tlc检测反应完全。慢慢倒入500g的冰水中充分搅拌静止分层,有机层用10%的碳酸氢钠水溶液洗,再加200g水洗之后减压脱溶得到淡黄色油状液体40g,gc含量96.8%。
如图5所示,在500毫升三口瓶中,在氮气保护下加入四氢呋喃200ml,新活化的镁条5g,滴加1ml的2,6-二乙基-4-甲基苄氯到反应瓶中,在室温下搅拌30分钟直到引发反应,降温到-5℃之后开始滴加40g的2,6-二乙基-4-甲基苄氯并控制在0℃左右2-3小时滴加完,滴完保温30分钟之后自然升到室温再保温1小时,用冰水降温到0℃通入干燥的二氧化碳气体,控制温度在10℃以下,直到反应不再放热过量二氧化碳气体溢出停止通气。加入200g水并用10%的稀盐酸调节ph值到5之后过滤出白色固体产品,烘干得到38.6g气相色谱分析含量98.4%。
如图6所示,将20g的2,6-二乙基-4-甲基苯甲酸固体投入100g的无水甲醇与10g甲苯磺酸中加热回流6小时,减压回收过量的甲醇之后加入100g冰水中充分搅拌,加50g乙酸乙酯萃取二次再合并有机层,浓缩回收乙酸乙酯之后得到20g的2,6-二乙基-4-甲基苯甲酸甲酯含量96.1%。
如图7所示,在500ml反应并中加入100ml的无水碳酸二甲酯通氮气保护,分批加入甲醇钠10.9g。加完升温回流,同时滴加20g的2,6-二乙基-4-甲基苯甲酸甲酯与50g碳酸二甲酯的混合液,滴加约3小时,加完保持回流6小时之后,取样检测原料反应完全,用冰水降温到0℃以下,滴加5%的稀盐酸,调节ph值到1-2静止分层得到有机层,减压回收溶济之后加10g甲醇重结晶得以20g的白色固体产品,含量97.3%。
本发明设计了一条全新的该化合物合成路线,避免了贵重金属催化剂的使用引起的成本昂贵的问题,在原料的选取和采用中杜绝了使用丙二腈高毒原料,提高了生产的安全性,有利于环保,各步反应条件温和收率稳定,安全性能高,有利于进行工业化生产。
最后应说明的是:以上所述仅为本发明的优选实施例而已,并不用于限制本发明,尽管参照前述实施例对本发明进行了详细的说明,对于本领域的技术人员来说,其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!