基于芳基并喹喔啉的D-A-D型环金属铱配合物近红外发光材料与应用的制作方法
本发明涉及近红外发光材料技术领域,具体涉及一类基于芳基并喹喔啉的d-a-d型环金属铱配合物近红外发光材料及应用。
背景技术:
近红外发光(700-2500nm),得益于极强的穿透力,在军事、通信、医疗和光疗领域显示出无与伦比的优势,目前,常用的近红外发光材料大致分为有机发光材料、无机发光材料、有机无机复合发光材料三大类。其中,有机近红外发光材料主要包括有机小分子、聚合物和金属配合物。根据发光机理的不同,又可以分为荧光材料和磷光材料。早期报导的近红外发光材料主要是稀土金属(如镱、铌、铒、铕等)配合物,其发光来源于跃迁禁阻的f-f跃迁,发光呈线状,但是该类近红外发光材料发光效率普遍较低,难以满足人们发展的需求。因此,有机小分子、聚合物和过渡金属配合物受到广大科研工作者的关注。不同于传统的有机小分子和聚合物近红外荧光材料,过渡金属(如锇、铱、铂)配合物可通过强烈的自旋轨道耦合作用,同时利用单、三线态激子发光,其内量子效率可达100%。目前,环金属铂配合物近红外发光材料的效率最高,外量子效率达24%。然而,铂(ii)配合物的平面四方构型、长的激发态寿命极易造成器件在高电流密度的激子湮灭和效率滚降(roll-off)。与环金属铂配合物不同,环金属铱(iii)配合物具有立体的八面体构型、相对较短的激发态寿命,可有效缓解有机近红外电致发光器件在高电流密度下的效率滚降问题。近红外环金属铱配合物近十年已取得长足的发展,在700-800nm波长范围的最大外量子效率达到4.5%。考虑到近红外电致发光材料及器件的重要性及目前效率不高的现状,设计开发一类简单、高效的近红外环金属铱配合物尤为重要。[1-9]
为此,我们发明了一类以芳基并喹喔啉为受电子(a)中心核,给电子(d)单元为外围基团的d-a-d型环金属配体及其环金属铱配合物。其中,环金属配体内核选用高度刚化的芳基并喹喔啉单元,以平衡发光波长和效率,打破能隙定律的限制,而外围给电子(d)单元可有效保护铱配合物发光核心、调节环金属铱配合物的聚集状态、溶解性、载流子传输能力并抑制器件效率在高电流密度下的滚降,提高近红外聚合物电致磷光器件的发光效率。
[1]wang,zy.crcpress/taylor&francisgroup,2013.
[2]qian,g;wang,zy.chemistryanasianjournal,2010,5,1006.
[3]xiang,h;cheng,j;ma,x;etal.chemicalsocietyreviews,2013,42,6128.
[6]liao,jl;chi,y;yeh,cc;etal.journalofmaterialschemistryc,2015,3,4910.
[7]cao,x;miao,j;zhu,m;etal.chemistryofmaterials,2015,27,96.
[8]ly,kt;chi,y;etal.naturephotonics,2017,11,63.
[9]xue,j;qiao,j;etal.chemistryofmaterials,2017,29,4775.
技术实现要素:
针对目前环金属铱(iii)配合物近红外发光材料发光效率普遍较低、构效关系不明确等问题,本发明开发了一类基于芳基并喹喔啉的d-a-d型环金属铱配合物及其在近红外聚合物电致发光二极管中的应用。此类d-a-d型环金属铱配合物具有以下结构特点:(1)d-a-d型环金属配体是以高度刚性的稠杂环化合物芳基并喹喔啉作为受电子(a)单元,富电子取代基作为推电子(d)单元,d-a-d构型的引入可大幅度调节环金属铱配合物的能隙,实现近红外发射;(2)d-a-d构型具有双极传输性能,可平衡电致发光器件中的载流子平衡,提高发光效率;(3)刚性稠杂环中心核a单元和外围立体d单元的协同作用,可以调节环金属铱配合物的聚集状态,提升环金属铱配合物的荧光量子效率,实现波长和效率的平衡,打破能隙定律的制约,实现高效近红外聚合物磷光器件。
本发明开发的d-a-d型环金属铱配合物合成步骤简单、产率高、易于纯化、并具有良好的溶解性和成膜性,可通过简单的溶液加工法制备高效近红外聚合物磷光器件。
此类基于芳基并喹喔啉的d-a-d型环金属铱配合物具有式1所示的化学结构:
其中:ar为多元取代芳环或芳杂环,d分别独立的选自氢原子、噻吩、三苯胺、咔唑、吖啶中的任何一种,辅助配体lx选自乙酰丙酮、二苯甲酰甲烷、吡啶羧酸,ar、d及lx的结构特征如式2所示:
其中,r为三氟甲基、碳原子数为1~16的直链或支链烷基、碳原子数为1~16的直链或支链烷氧基。
优选的方案,此类基于芳基并喹喔啉的d-a-d型环金属铱配合物,包括下列式3中基于芳基并喹喔啉的d-a-d型环金属配体的任何衍生物所形成的环金属铱(iii)配合物近红外发光材料。
其中,r为三氟甲基、碳原子数为1~16的直链或支链烷基、碳原子数为1~16的直链或支链烷氧基。
为了获得上述基于芳基并喹喔啉的d-a-d型环金属铱配合物,本发明优选的合成方案如下:通过suzuki偶联反应、still偶联反应、schiff碱缩合反应获得相应的d-a-d构型的环金属配体,再通过桥连及去桥连反应获得d-a-d型环金属铱配合物。
本发明还提供此类基于芳基并喹喔啉的d-a-d型环金属铱配合物在聚合物电致发光器件中的应用,将其作为单一活性发光材料应用于聚合物电致发光器件的发光层,制备高效近红外发光的聚合物电致发光器件。
近红外聚合物电致发光器件的结构包括氧化铟锡(ito)导电玻璃衬底阳极,空穴注入层,发光层,电子传输层和阴极。其中,空穴注入层为聚二氧乙基噻吩以及聚苯乙烯磺酸(pedot:pss)涂层,空穴传输层为聚[双(4-苯基)(4-丁基苯基)胺](poly-tpd)涂层,发光层为单一发光材料和主体材料的共混涂层或者不掺主体材料的单一发光材料涂层,1,3,5-三(1-苯基-1h-苯并咪唑-2-基)苯(tpbi)为电子传输层,氟化铯(csf)为电子注入层,阴极为铝的沉积层。
主体材料为4,4-二(9-咔唑)联苯(cbp),发光层中发光材料与主体材料的质量百分比分别为1~100%,99~0%。
附图说明
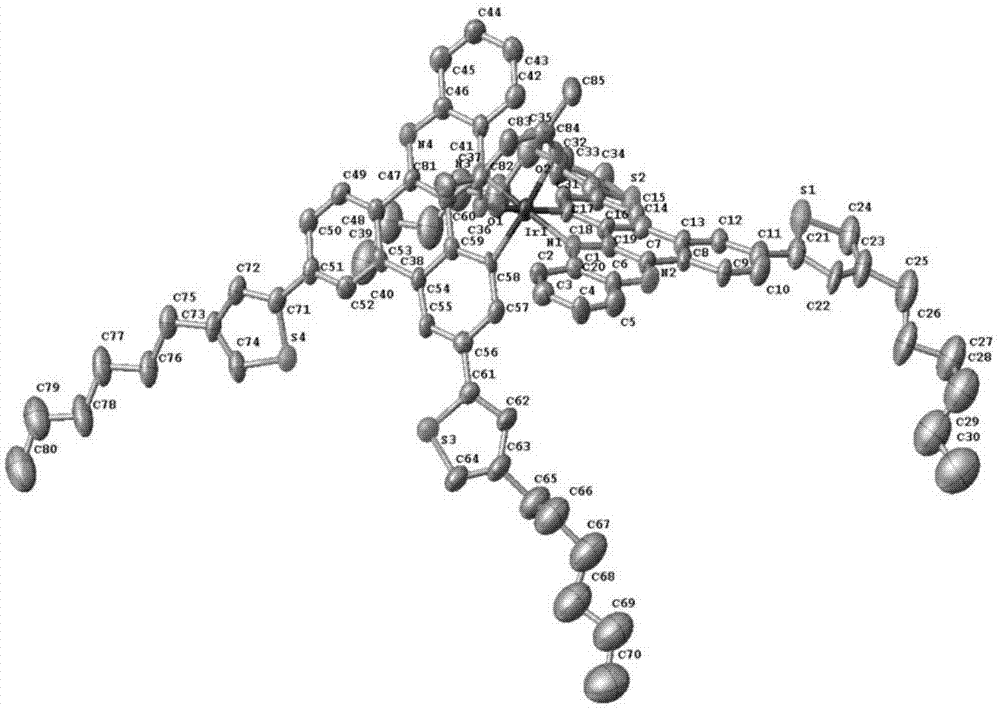
图1为本发明实施例1中配合物ir-1的单晶结构图。
图2为本发明实施例1中配合物ir-1的晶体堆积图。
图3为本发明实施例1中配合物ir-1的紫外-可见吸收光谱图。
图4为本发明实施例1中配合物ir-1的光致发光光谱图。
图5为本发明实施例2中配合物ir-2的紫外-可见吸收光谱图。
图6为本发明实施例2中配合物ir-2的光致发光光谱图。
图7为本发明实施例3中配合物ir-3的紫外-可见吸收光谱图。
图8为本发明实施例3中配合物ir-3的光致发光光谱图。
图9为本发明实施例4中配合物ir-4的紫外-可见吸收光谱图。
图10为本发明实施例4中配合物ir-4的光致发光光谱图。
图11本发明实施例1中配合物ir-1的电致发光光谱图(el)。
图12本发明实施例1中配合物ir-1的外量子效率-电流密度曲线(eqe-j)图。
图13本发明实施例2中配合物ir-2的电致发光光谱图(el)。
图14本发明实施例2中配合物ir-2的外量子效率-电流密度曲线(eqe-j)图
图15本发明实施例3中配合物ir-3的电致发光光谱图(el)。
图16本发明实施例3中配合物ir-3的外量子效率-电流密度曲线(eqe-j)图
图17本发明实施例4中配合物ir-4的电致发光光谱图(el)。
图18本发明实施例4中配合物ir-4的外量子效率-电流密度曲线(eqe-j)图
具体实施方案
以下具体实施例旨在对本发明作进一步说明,但这些具体实施方案不以任何方式限制本发明的保护范围。
实施例1
基于芳基并喹喔啉的d-a-d型环金属铱配合物ir-1的制备
铱配合物ir-1的合成路线如下:
中间体1的合成
在100ml单口瓶中,依次加入1,2-苯二胺(325mg,3.00mmol)、3,6-二溴-9,10-菲醌(1g,2.73mmol)和无水乙醇(30ml)、醋酸(1ml),加热80℃,回流搅拌4h,析出大量黄绿色固体。冷却至室温、抽滤,滤饼经无水乙醇(10×3ml)洗涤后真空干燥3h,得黄绿色固体1.19g,产率91%,直接投入下一步反应。
中间体2的合成
在100ml双口瓶中,依次加入中间体1(500g,1.14mmol)、(4-己基噻吩基)三丁基锡(1.56g,3.42mmol)、四(三苯基膦)合钯(40mg,0.034mmol)、甲苯(20ml),氮气氛围下,加热至80℃,搅拌反应12h。减压蒸馏除去甲苯,剩余物用二氯甲烷萃取、水洗、无水mgso4干燥、过滤,滤液经减压蒸馏除去溶剂,剩余物经硅胶柱色谱分离纯化,洗脱剂为二氯甲烷和石油醚的混合溶液(1/3,v/v),得亮黄色固体558mg,产率80%。1hnmr(400mhz,cdcl3)δ(ppm):9.29(d,j=8.4hz,2h),8.67(d,j=1.3hz,2h),8.26(dd,j=6.5,3.4hz,2h),7.92(dd,j=8.4,1.5hz,2h),7.81(dd,j=6.5,3.4hz,2h),7.42(d,j=1.0hz,2h),7.02(s,2h),2.75–2.67(m,4h),1.73(dt,j=15.4,7.5hz,4h),1.43(dd,j=14.7,6.4hz,4h),1.37(dt,j=7.6,3.8hz,8h),0.93(t,j=7.0hz,6h).maldi-tof-ms(m/z):calcdforc40h40n2s2:612.89;found,614.26[m+1]+.
铱配合物ir-1的合成
在100ml单口瓶中,加入中间体2(200mg,0.33mmol)、1,5-环辛二烯氯化铱二聚体(54mg,0.08mmol)、甲苯(20ml),氮气保护下,加热至115℃。搅拌36h后停止反应,减压蒸馏除去甲苯,真空干燥3h,得黑色氯桥连体110mg,产率50%,未经纯化直接投入下一步反应。
上述氯桥连体(110mg,0.04mmol),tbuok(22mg,0.20mmol)、乙酰丙酮(20mg,0.20mmol)、30ml二氯甲烷与甲醇的混合溶剂(7/3,v/v)依次加入到100ml单口瓶中,氮气保护下,40℃搅拌过夜,冷却至室温后,蒸馏水洗涤,二氯甲烷萃取(20×2ml),无水mgso4干燥3h,过滤,滤液经减压蒸馏除去溶剂。剩余物以二氯甲烷/石油醚(1/3,v/v)为洗脱剂,通过硅胶柱色谱分离纯化,得黑色固体48mg,产率40%。1hnmr(400mhz,cdcl3)δ(ppm):9.40(d,j=8.4hz,2h),8.75(d,j=8.9hz,2h),8.69(s,2h),8.45(d,j=8.4hz,2h),8.17(s,2h),8.00(d,j=8.2hz,2h),7.89–7.81(m,2h),7.74–7.63(m,2h),7.46(s,2h),7.02(s,2h),6.80(s,2h),6.69(d,j=6.1hz,4h),4.90(s,1h),2.70(t,j=7.6hz,4h),2.41(t,j=7.6hz,4h),1.71(dd,j=14.8,7.4hz,4h),1.59(s,4h),1.49–1.38(m,8h),1.35(d,j=3.7hz,8h),1.25(s,14h),0.91(t,j=6.9hz,6h),0.84(t,j=6.7hz,6h).13cnmr(126mhz,cdcl3)δ(ppm):186.24,156.44,150.76,144.75,144.23,144.04,144.02,143.55,142.99,142.72,141.05,137.02,136.05,133.94,133.60,131.05,130.91,130.06,129.04,128.87,126.66,125.86,125.64,125.50,125.06,120.78,120.31,119.97,113.24,101.06,77.27,77.01,76.76,31.72,31.62,30.69,30.48,30.42,30.18,29.09,28.87,28.35,22.65,22.56,14.13,14.09,1.03.maldi-tof-ms(m/z):calcdfor[m-acac]+c80h78irn4s4:1415.99;found,1415.01.
实施例2
基于芳基并喹喔啉的d-a-d型环金属铱配合物ir-2的制备
铱配合物ir-2的合成路线如下:
中间体3的合成
在100ml双口瓶中,依次加入实施例1中的中间体1(500g,1.14mmol)、4-硼酸三苯胺(824mg,2.85mmol)、四(三苯基膦)合钯(40mg,0.034mmol)、2mol/l碳酸钾溶液(5ml)、无水乙醇(1ml)、甲苯(25ml),氮气氛围下,加热至80℃,搅拌反应12h。减压蒸馏除去甲苯,剩余液用二氯甲烷萃取、水洗、无水mgso4干燥、过滤,滤液经减压蒸馏除去溶剂,剩余物以二氯甲烷和石油醚的混合溶液(1/2,v/v)为洗脱剂,通过硅胶柱分离纯化,得黄色固体716mg,产率82%。1hnmr(400mhz,cdcl3)δ(ppm):9.41(dd,j=8.3,3.1hz,2h),8.76(s,2h),8.36–8.26(m,2h),7.95(d,j=6.3hz,2h),7.88–7.81(m,2h),7.75–7.63(m,4h),7.36–7.27(m,8h),7.25(d,j=3.2hz,4h),7.19(d,j=5.4hz,8h),7.07(s,4h).maldi-tof-ms(m/z):calcdforc56h38n4:766.95;found,767.49[m+1]+.
铱配合物ir-2的合成
合成方法同实施例1中铱配合物ir-1的合成,得黑褐色固体60mg,产率25%。1hnmr(400mhz,cdcl3)δ(ppm):9.45(d,j=8.4hz,2h),8.78(d,j=8.7hz,2h),8.71(s,2h),8.43(d,j=8.4hz,2h),8.16(s,2h),7.99(d,j=8.4hz,2h),7.84(t,j=7.6hz,2h),7.72(d,j=8.6hz,4h),7.68–7.61(m,2h),7.30(t,j=7.8hz,8h),7.25–7.14(m,20h),7.06(dd,j=11.5,8.0hz,8h),6.97(dd,j=13.6,7.5hz,12h),6.85(d,j=8.5hz,4h),6.78(s,2h),4.88(s,1h),1.58(s,6h).13cnmr(126mhz,cdcl3)δ(ppm):186.19,156.72,151.00,147.92,147.54,147.51,147.15,144.23,143.12,143.00,142.66,142.40,140.51,135.41,134.38,134.14,133.78,132.10,130.64,130.12,129.36,129.18,128.36,128.26,126.57,126.38,125.07,124.64,124.38,123.69,123.43,123.20,122.83,121.08,114.20,77.27,77.01,76.76,29.71,28.35.maldi-tof-ms(m/z):calcdfor[m-acac]+c112h74irn8:1724.10;found,1723.46.
实施例3
基于芳基并喹喔啉的d-a-d型环金属铱配合物ir-3的制备
铱配合物ir-3的合成路线如下:
中间体4的合成
在100ml双口瓶中,依次加入4-硼酸三苯胺(1.40g,4.70mmol)、4,5-二溴邻苯二胺(0.50g,1.88mmol)、四(三苯基膦)合钯(130mg,0.029mmol)、2mol/l碳酸钾溶液(8ml)、甲苯(20ml),氮气氛围下,加热至80℃,搅拌反应12h。减压蒸馏除去甲苯,剩余液用二氯甲烷萃取、水洗、无水mgso4干燥、过滤,滤液经减压蒸馏除去溶剂,真空干燥3h,得白色固体894mg,产率80%。因产物对氧气极其敏感,故未经纯化直接投入下一步反应。
中间体5的合成
在100ml单口瓶中,依次加入中间体4(894mg,1.50mmol)、9,10-菲醌(312mg,1.50mmol)和无水乙醇(30ml)、醋酸(1ml),80℃回流搅拌4h,析出大量橙色固体,冷却至室温、抽滤、滤饼用无水乙醇(10×3ml)洗涤,以二氯甲烷和石油醚的混合溶液(1/1,v/v)为洗脱剂,通过硅胶柱分离纯化,得橙色固体1g,产率87%。1hnmr(300mhz,cdcl3)δ(ppm):9.41(dd,j=7.8,1.6hz,2h),8.61–8.55(m,2h),8.36(s,2h),7.83–7.72(m,4h),7.29(dd,j=6.9,1.5hz,6h),7.25–7.19(m,6h),7.14(dd,j=8.5,1.1hz,8h),7.08–7.01(m,8h).maldi-tof-ms(m/z):calcdforc56h38n4:766.95;found,767.49[m+1]+.
铱配合物ir-3的合成
合成方法同实施例1中铱配合物ir-1的合成,得黑色固体78mg,产率33%。1hnmr(400mhz,cdcl3)δ(ppm):9.43(d,j=7.7hz,2h),8.82(s,2h),8.48(d,j=3.8hz,4h),7.93(d,j=7.9hz,2h),7.81–7.73(m,4h),7.31–7.26(m,12h),7.17–7.10(m,16h),7.05(dd,j=7.9,5.0hz,8h),6.96(ddd,j=20.1,10.4,5.5hz,18h),6.85(d,j=8.6hz,4h),6.62(d,j=7.5hz,2h),4.88(s,1h),1.14(s,6h).13cnmr(126mhz,cdcl3)δ(ppm):186.58,156.71,150.14,147.58,147.49,147.06,146.88,144.39,143.99,142.30,142.26,141.34,134.49,134.01,133.85,133.28,132.85,131.01,130.89,130.65,130.29,130.03,129.93,129.33,129.14,127.60,125.97,124.62,124.23,123.23,123.09,123.06,122.79,122.60,114.88,101.51,77.26,77.01,76.76,29.70,27.90.maldi-tof-ms(m/z):calcdfor[m-acac]+c112h74irn8:1724.10;found,1724.11.
实施例4
基于芳基并喹喔啉的d-a-d型环金属铱配合物ir-4的制备
铱配合物ir-4的合成路线如下:
中间体6的合成
合成方法同实施例3中中间体5的合成,得橙红色固体1.13g,产率95%。1hnmr(300mhz,cdcl3)δ(ppm):9.59(dd,j=7.7,1.1hz,1h),8.41(s,1h),8.29(dd,j=7.7,1.1hz,1h),8.09(dd,j=15.3,7.6hz,2h),7.29(d,j=8.3hz,3h),7.23(s,3h),7.17–7.13(m,4h),7.08–7.04(m,4h).maldi-tof-ms(m/z):calcdforc58h38n4:790.79;found,791.56[m+1]+.
铱配合物ir-4的合成
合成方法同实施例1中铱配合物ir-1的合成,得黑色固体101mg,产率43%。1hnmr(400mhz,cdcl3)δ(ppm):9.55(d,j=7.7hz,2h),8.82(s,2h),8.52(s,2h),8.24(d,j=7.5hz,2h),8.02(t,j=7.8hz,2h),7.78(q,j=8.9hz,4h),7.44(d,j=8.2hz,2h),7.28(dd,j=8.8,7.1hz,10h),7.18–7.03(m,28h),6.98(d,j=8.6hz,4h),6.94–6.87(m,12h),6.82(d,j=8.6hz,4h),4.93(s,1h),1.17(s,6h).13cnmr(126mhz,cdcl3)δ(ppm):186.64,158.47,151.81,147.58,147.43,147.07,146.89,145.28,144.57,142.65,142.40,142.19,139.86,134.40,134.01,132.74,131.92,131.00,130.89,130.43,129.89,129.34,129.12,128.72,127.94,127.62,126.79,126.71,125.58,124.64,124.22,123.64,123.11,122.99,122.78,122.59,122.50,101.61,77.26,77.01,76.76,29.70,27.88.maldi-tof-ms(m/z):calcdfor[m-acac]+c116h74irn8:1772.14;found,1772.83.
实施例5
实施例1中环金属铱配合物ir-1的x-射线单晶衍射实验
通过溶剂扩散法获得环金属铱配合物ir-1的单晶,具体为无水甲醇向饱和二氯甲烷溶液中挥发缓慢生长单晶。其x-射线单晶衍射结果如附图1所示。实验结果表明,环金属铱配合物ir-1呈现经典的八面体构型,两个环金属配体和辅助配体围绕在中心铱原子周围。环金属配体呈现c,c顺位,n,n反位的构型。铱原子与环金属配体的配位键长为
实施例6
环金属铱配合物ir-1的光物理性能
配合物ir-1的紫外-可见(uv-vis)吸收光谱及荧光光谱(pl)分别如附图3和图4所示。将铱配合物ir-1配制成1×10-5mol/l的二氯甲烷(dcm)溶液,并移取3ml置于石英比色皿中,测试其uv-vis和pl光谱;配制10mg/ml的dcm溶液,通过旋转涂敷于石英片上,测试薄膜的uv-vis和pl光谱。
由图3可以看出:(1)配合物ir-1在250nm至700nm均有吸收并呈现三个特征吸收带,其中,250nm到370nm的强吸收带归属于环金属配体的π→π*电荷转移跃迁吸收,而370nm至550nm的中强吸收带归属于配体内的电荷转移跃迁吸收(ict)和自旋允许的金属到配体的电荷转移跃迁吸收(1mlct),500nm至700nm较弱的吸收来源于自旋禁阻的3mlct和配体到配体的电荷转移跃迁吸收(3llct);(2)在固体薄膜中,配合物ir-1的吸收峰呈现明显的红移,说明在固体薄膜中,存在较强的分子间相互作用。
由图3可以看出:配合物ir-1不论在二氯甲烷溶液还是固体薄膜中均呈现强烈的近红外发射,其在二氯甲烷中的最大发射峰为712nm,而在固体薄膜中,除了710nm左右的发射峰外,在801nm附近又出现一个新的发射峰,说明在固体薄膜中分子间的相互作用特别强,呈现出明显的激基缔合物发射,这为构筑近红外发光铱配合物提供了新途径。
实施例7
环金属铱配合物ir-2的光物理性能
配合物ir-2的紫外-可见(uv-vis)吸收光谱及荧光光谱(pl)分别如附图5和图6所示。由图5可以看出,配合物ir-2在250nm至700nm均有吸收并呈现两个特征吸收带,其中250nm至400nm的强吸收归属于配体内的π→π*电荷转移跃迁吸收,400nm一直延伸至700nm的吸收归属于配体内的电荷转移跃迁吸收(ict)和自旋允许的1mlct以及自旋禁阻的3mlct、3llct混合吸收。在固体薄膜中,477nm附近的吸收峰相对溶液的460nm有17nm红移,表明配合物ir-2在固体薄膜中存在较强的分子间相互作用。
由图6的pl光谱可以看出,配合物ir-2在二氯甲烷溶液中呈现宽而无精细结构的近红外发射,表明主要来源于mlct的发射,其最大发射峰位于717nm,而在固体薄膜中时表现出明显的激基缔合物发射,呈现两个发射峰,分别位于701和790nm,且790nm的激基缔合物发射占主导位置。
实施例8
环金属铱配合物ir-3的光物理性能
配合物ir-3的紫外-可见(uv-vis)吸收光谱及荧光光谱(pl)分别如附图7和图8所示。由图7可以看出:(1)配合物ir-3在200nm至700nm均有吸收并呈现三个特征吸收带,其中,200nm到400nm的强吸收带归属于环金属配体的π→π*电荷转移跃迁吸收;400nm至550nm的中强吸收带归属于配体内的电荷转移跃迁吸收(ict)和自旋允许的金属到配体的电荷转移跃迁吸收(1mlct);500nm至700nm较弱的吸收来源于自旋禁阻的3mlct和配体到配体的电荷转移跃迁吸收(3llct);(2)在固体薄膜中,配合物ir-3的吸收峰呈现明显的红移,在溶液中位于492nm的吸收峰红移至薄膜中的500nm,发生8nm的红移,说明在固体薄膜中,存在分子间相互作用。
由图8的pl光谱可以看出,配合物ir-3在二氯甲烷溶液中呈现宽而无精细结构的近红外发射,表明主要来源于mlct的发射,其最大发射峰位于727nm,而在固体薄膜中时呈现出与二氯甲烷中一致的发射光谱,其最大发射峰位于726nm,说明配合物ir-3在薄膜中无明显的分子间相互作用,发射光谱的蓝移表明在固体薄膜中分子内的扭曲、振动受到抑制,辐射跃迁速率增加所致。
实施例9
环金属铱配合物ir-4的光物理性能
配合物ir-4的紫外-可见(uv-vis)吸收光谱及荧光光谱(pl)分别如附图9和图10所示。由紫外-可见吸收图9可以看出:(1)配合物ir-4在200nm至750nm均有吸收并呈现三个特征吸收带,其中,200nm到450nm的强吸收带归属于环金属配体的π→π*电荷转移跃迁吸收,450nm至550nm的中强吸收带归属于配体内的电荷转移跃迁吸收(ict)和自旋允许的金属到配体的电荷转移跃迁吸收(1mlct),550nm至750nm较弱的吸收来源于自旋禁阻的3mlct和配体到配体的电荷转移跃迁吸收(3llct);(2)在固体薄膜中,配合物ir-4的吸收峰呈现明显的红移,在溶液中位于504nm的吸收峰,在薄膜中红移至521nm,发生了17nm的红移,说明在固体薄膜中,存在分子间相互作用。
由图10的pl光谱可以看出,配合物ir-4在二氯甲烷溶液中呈现宽而无精细结构的近红外发光,表明主要来源于mlct的发射,其最大发射峰位于772nm,而在固体薄膜中时其最大发射峰位于759nm,说明配合物ir-4在薄膜中无明显的分子间相互作用,发射光谱的蓝移表明在固体薄膜中分子内的扭曲、振动受到抑制,辐射跃迁速率增加所致。
实施例10
环金属铱配合物ir-1的聚合物电致发光器件性能表征
聚合物电致发光器件的掺杂器件结构为ito(110nm)/pedot:pss(40nm)/poly-tpd(30nm)/cbp:ir-1,50nm)/tpbi(60nm)/csf(0.8nm)/al(100nm);非掺杂器件结构为ito(110nm)/pedot:pss(40nm)/pvk(30nm)/ir-1(50nm)/tpbi(60nm)/csf(0.8nm)/al(100nm)。其中,氧化铟锡(ito)玻璃基底为阳极,聚二氧乙基噻吩(pedot)和聚苯乙烯磺酸钠(pss)为空穴注入层,聚[双(4-苯基)(4-丁基苯基)胺](poly-tpd)和聚乙烯基咔唑(pvk)分别为空穴传输层,1,3,5-三(1-苯基-1h-苯并咪唑-2-基)苯(tpbi)为电子传输层,氟化铯(csf)为电子注入层,阴极为铝的沉积层。在掺杂器件中,主体材料是4,4-二(9-咔唑)联苯(cbp),发光层是主体材料和配合物客体材料(dopant)ir-1的共混涂层,掺杂配合物的质量百分数为1%、2%、4%、8%、50%,在非掺杂器件中,发光层为单一的配合物涂层。
聚合物电致发光器件的电致发光光谱图(el)、外量子效率-电流密度曲线(eqe-j)分别如附图11和12所示。由图可见:在掺杂质量百分数为4%的器件中,器件的最大电致发光峰位于708nm,eqemax达到2.43%,最大辐照度(rmax)为1125μw/sr/cm2。在掺杂质量百分数为50%时,ir-1器件呈现两个发光峰,分别位于718和778nm,其eqemax为1.35%,表明随着掺杂浓度的提高,配合物ir-1分子间的相互作用逐渐增强,表现出单体发射和激基缔合物的发射。在非掺杂的器件中,激基缔合物的发射峰更加明显,但同时也存在单体的发射,发射峰分别位于714和782nm,与pl光谱的发光峰吻合,表明是来自配合物ir-1的本征发射,eqemax达到0.35%,最大辐照度(rmax)为136μw/sr/cm2。值得注意的是,以配合物ir-1为发光层的聚合物电致发光器件都呈现出较小的效率滚降,表明配合物ir-1是一种性能优良的近红外磷光材料。
实施例11
环金属铱配合物ir-2的聚合物近红外电致发光器件性能表征
聚合物电致发光器件的器件结构与实施例10相同。聚合物电致发光器件的电致发光光谱图(el)、外量子效率-电流密度曲线(eqe-j)分别如附图13和14所示。由图可见:在掺杂质量百分数为2%的器件中,器件性能最佳,最大电致发光峰位于702nm,eqemax达到6.05%,最大辐照度(rmax)为948μw/sr/cm2。在非掺杂的器件中,呈现激基缔合物的发射峰,但ir-2单体的发射峰占主导位置,发射峰分别位于714和780nm,eqemax达到0.47%,最大辐照度(rmax)为88μw/sr/cm2。值得注意的是,以配合物ir-2为发光层的聚合物电致发光器件都呈现出较小的效率滚降,表明配合物ir-2是一种性能优良的近红外磷光材料。
实施例12
环金属铱配合物ir-3的聚合物近红外电致发光器件性能表征
聚合物电致发光器件的器件结构与实施例10相同,掺杂配合物ir-3的质量百分数为1%、2%、4%、8%。聚合物电致发光器件的电致发光光谱图(el)、外量子效率-电流密度曲线(eqe-j)分别如附图15和16所示。由图可见:在掺杂质量百分数为1%的器件中,器件性能最佳,最大电致发光峰位于712nm,eqemax达到6.22%,最大辐照度(rmax)为1416μw/sr/cm2。在掺杂质量百分数为8%的器件中,最大电致发光峰位于724nm,eqemax为3.47%,最大辐照度(rmax)为1296μw/sr/cm2。值得注意的是,以配合物ir-3为发光层的聚合物电致发光器件都呈现出较小的效率滚降和浓度猝灭,表明配合物ir-3是一种性能优良的近红外磷光材料。
实施例13
环金属铱配合物ir-4的聚合物近红外电致发光器件性能表征
聚合物电致发光器件的器件结构与实施例10相同,掺杂配合物ir-4的质量百分数为1%、2%、4%、8%。聚合物电致发光器件的电致发光光谱图(el)、外量子效率-电流密度曲线(eqe-j)分别如附图17和18所示。由图可见:在掺杂质量百分数为2%的器件中,器件性能最佳,最大电致发光峰位于772nm,eqemax达到2.77%,最大辐照度(rmax)为791μw/sr/cm2。在掺杂质量百分数为8%的器件中,最大电致发光峰位于780nm,eqemax为1.62%,最大辐照度(rmax)为521μw/sr/cm2。值得注意的是,以配合物ir-4为发光层的聚合物电致发光器件都呈现出较小的效率滚降和浓度猝灭,表明配合物ir-4是一种性能优良的近红外磷光材料。
尽管结合了优选实施例对本发明进行了说明,但本发明并不局限于上述实施案例,应当理解所附权利要求概括了本发明的范围。在本发明构思的指导下,本领域的技术人员应当意识到,对本发明的各实施例方案所进行的一定的改变,都将被本发明的权利要求书的精神和范围所覆盖。
- 还没有人留言评论。精彩留言会获得点赞!