一种N’-(4-烷苯基)-N,N-二甲基脒的合成分离方法与流程
本发明属于有机合成技术领域,更具体的,涉及一种n’-(4-烷苯基)-n,n-二甲基脒的合成分离方法。
背景技术:
n’-烷苯基脒类化合物可用作消炎、抗菌药,还一些种类n’-烷苯基脒类化合物具有较好的抗癌特性。在农业上广泛作为杀虫剂,具有低毒、高活、环保的优点。n’-烷苯基脒类化合物还可以作为有机合成的中间体,用于含有氮杂环类化合物的合成。n’-长链烷苯基脒作为co2/n2开关表面活性剂,在破乳性能方面有较大优势。通常情况下,在co2开关型表面活性剂体系的质子化及去质子化过程中,去质子化更难进行,所述去质子化即表面活性剂失去表面活性从而乳液破乳的过程。端基的碱性程度会影响体系去质子化反应的平衡。研究表明,端基碱性越低,去质子化过程更易进行。与烷基脒类表面活性剂相比,烷苯基脒类端基具有更低的碱性,因而其去质子化过程会更加迅速和彻底。与n’-十二烷基-n,n-二甲基乙脒/水体系的去质子化能力相比,在室温下向表面活性剂体系中通入ar,烷苯基脒碳酸氢盐/水体系电导率迅速下降,在短时间内基本完成去质子化过程。而十二烷基乙脒碳酸氢盐/水体系在相同的时间内,电导率仅下降了不到20%,大部分乙脒仍是质子化状态(fowlerci,jessoppg,cunninghammf.arylamidineandtertiaryamineswitchablesurfactantsandtheirapplicationintheemulsionpolymerizationofmethylmethacrylate[j].macromolecules,2012,45:2955-2962.)。
目前,公开报道的n’-烷苯基脒类的合成多采用以伯胺与n,n-二甲基乙酰胺二甲基缩醛为原料的酰胺缩醛法(jitendrar.harjani,chenl,jessoppg.asynthesisofacetamidines[j].thejournaloforganicchemistry,2011,76:1683-1691)。
该方法可采用以下三种方式:
(1)将伯胺(3.18mmol)与n,n-二甲基乙酰胺二甲基缩醛(3.82mmol)混合,在60℃下,加热2h。反应混合物在旋转蒸发器上室温下旋蒸,高真空条件下,55℃继续加热8h,得到产物,固体胺用thf(5ml)为溶剂;
(2)向n,n-二甲基乙酰胺二甲基缩醛(3.82mmol)中逐滴加入伯胺(3.18mmol),滴加时间在5分钟以上,与此同时室温下搅拌10分钟。室温下反应18h后,使用旋转蒸发仪去除挥发性组分。之后在55℃高真空下继续加热8h,以获得产品。对于固体胺,以thf(5ml)为溶剂,在胺的thf溶液中逐滴滴加n,n-二甲基乙酰胺二甲基缩醛;
(3)n,n-二甲基乙酰胺二甲基缩醛(3.82mmol)和me2nh(3.8ml,2m溶解在thf,7.64mmol)在圆形底瓶中混合,室温下搅拌。在室温条件下,逐滴加入癸胺(3.18mmol),滴加时间5分钟以上。搅拌10分钟,避光反应18小时。之后旋蒸除去thf和me2nh。然后在高真空下55℃搅拌8h。
fowler等,在圆底烧瓶中,4-癸基苯胺(4.2mol)与n,n-二甲基乙酰胺二甲基缩醛(4.2mmol)在60℃下反应30min,反应液通过旋转蒸发除去甲醇后,得到n’-(4-癸苯基)-n,n-二甲基乙脒(fowlerci,jessoppg,cunninghammf.arylamidineandtertiaryamineswitchablesurfactantsandtheirapplicationintheemulsionpolymerizationofmethylmethacrylate[j].macromolecules,2012,45:2955-2962.)。
上述方法分两个阶段进行。第一阶段所需时间,因反应温度不同而不同。室温下反应约需18小时;60℃下反应需要30min~120min。第二阶段则在高真空条件下,抽除反应生成的醇和加入的溶剂,该阶段需持续操作约8小时,以所获较高纯度的产品。该方法合成过程所需时间较长,而且有时需要加入一定量的溶剂(如,四氢呋喃)溶解烷胺或烷基苯胺,溶剂将在第二阶段除去。
综上所述,目前需要提供一种n’-(4-烷苯基)-n,n-二甲基脒的高效合成方法,在得到高纯度的最终产品的前提下,缩短合成时间。
技术实现要素:
为了解决上述问题,本发明提供一种n’-(4-烷苯基)-n,n-二甲基脒的合成分离方法,该方法集反应与分离同时进行,缩短了合成时间、提高了合成效率。
为了实现以上目的,本发明采用以下技术方案:
一种n’-(4-烷苯基)-n,n-二甲基脒的合成分离方法,该方法的反应式为:
其中,r1为c12-c18的烷基,r2为h或-ch3。
具体的,该方法包括以下步骤:
将4-烷基苯胺溶于乙腈中,加热至预设温度后,加入n,n-二甲基酰胺二甲基缩醛进行反应;
反应结束后将反应体系冷却,待所述反应体系分层后,取下层液体减压蒸馏,即得到产品n’-(4-烷苯基)-n,n-二甲基脒。
反应体系分层可直接静置分层也可通过离心等手段分层。上层液体为乙腈、未反应的4-烷基苯胺和缩醛;将上层清液在压力1kpa-100kpa,温度30℃-100℃减压蒸馏回收乙腈,回收的乙腈可以循环使用1-10次;优选地,使用次数为5-7次。
本发明的方法在酰胺缩醛法的基础上,引入乙腈作为溶剂。乙腈作为溶解原料4-烷基苯胺的溶剂的同时,作为反应分离过程的萃取剂,利用反应原料与产物n’-(4-烷苯基)-n,n-二甲基脒在乙腈中的溶解度差异,在反应过程中实现产物与反应物的分离。反应结束后,待反应液冷却静置分层后,即可获得含有少量溶剂的n’-(4-烷苯基)-n,n-二甲基脒产物,通过减压蒸馏处理,可迅速获得高纯度的n’-(4-烷苯基)-n,n-二甲基脒。而含有未反应完全的原料的乙腈,可通过减压精馏,回收利用。该方法是一种通过溶液萃取过程强化的化学反应,实现了n’-(4-烷苯基)-n,n-二甲基脒的高效合成方法。
在以上方法中,优选地,所述乙腈与4-烷基苯胺的体积比为5:1-20:1。
在以上方法中,将4-烷基苯胺溶于乙腈中,加热至预设温度以使乙腈回流,以加快反应进程,缩短反应时间。优选地,所述预设温度为50-90℃。
在以上方法中,需采取n,n-二甲基酰胺二甲基缩醛与4-烷基苯胺的反应原料比高于反应化学计量比的方案,以使烷基苯胺尽可能反应完全。优选地,所述n,n-二甲基酰胺二甲基缩醛与4-烷基苯胺的摩尔比为1:1-2:1。
优选地,所述反应为常压反应10min-20min。优选地,所述减压蒸馏的压力为1kpa-100kpa,温度为30℃-100℃。
本发明的n’-(4-烷苯基)-n,n-二甲基脒的合成法中,采用的缩醛优选地为n,n-二甲基甲酰胺二甲基缩醛和n,n-二甲基乙酰胺二甲基缩醛中的一种或两种组合。
优选地,所述烷基苯胺为4-烷基苯胺的烷基为c12-c18的烷基。
本发明另一方面提供上述方法制备出的n’-(4-烷苯基)-n,n-二甲基脒,其结构式如下:
其中r1为c12-c18烷基,r2为h或-ch3。
本发明的方法采用乙腈作为反应溶剂和萃取剂,利用反应组分4-烷基苯胺、n,n-二甲基酰胺缩醛及n’-(4-烷苯基)-n,n-二甲基脒在乙腈中溶解度差异,实现反应与萃取分离的耦合,获得高纯度的n’-(4-烷苯基)-n,n-二甲基脒。该方法所获得的n’-(4-烷苯基)-n,n-二甲基脒的收率可达到90%以上,萃取分离后产品n’-(4-烷苯基)-n,n-二甲基脒的纯度可进一步提高至95%及以上。并有效缩短操作时间,提高合成效率,本方法反应分离操作简单,萃取溶剂可循环使用。本发明的n’-(4-烷苯基)-n,n-二甲基脒可以用农药、医用药物,也是一种新型co2/n2开关表面活性剂。
附图说明
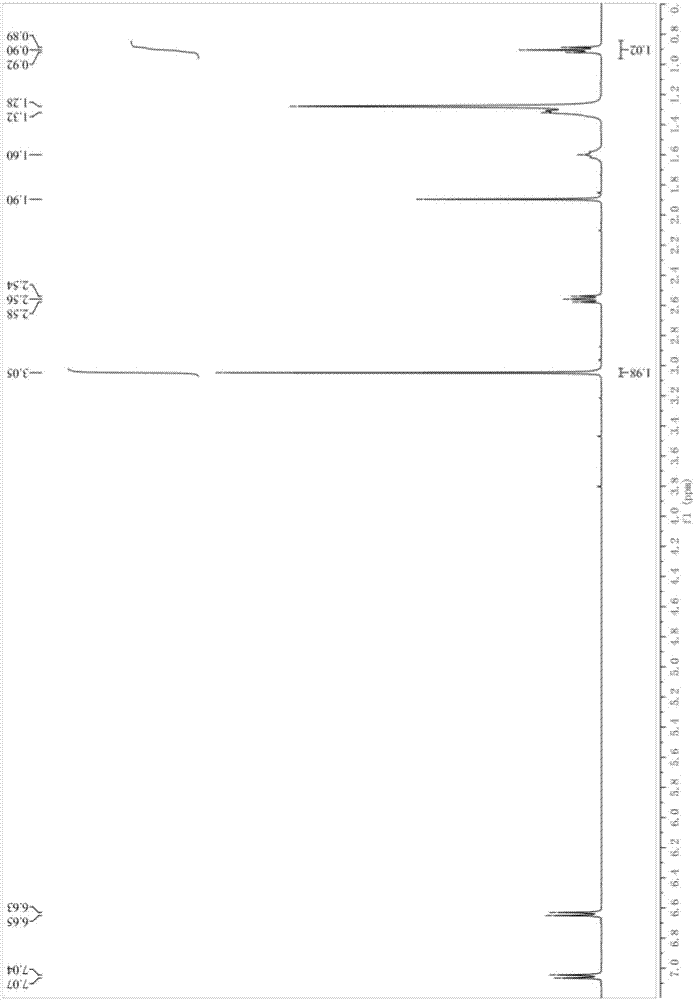
图1为实施例1中制备的n’-(4-十四烷苯基)-n,n-二甲基乙脒的1hnmr(cdcl3,500m)谱图。
图2为实施例1中制备的n’-(4-十四烷苯基)-n,n-二甲基乙脒的红外谱图。
具体实施方式
为了对本发明的技术特征、目的和有益效果有更加清楚的理解,现对本发明的技术方案进行以下详细说明,但不能理解为对本发明的可实施范围的限定。
本发明实施例中所涉及到的化学试剂如无特殊说明均可商业购买获得。
实施例1
本实施例提供了一种n’-(4-十四烷苯基)-n,n-二甲基乙脒,通过以下步骤合成得到:
将5mmol的4-十四烷基苯胺加入带有回流装置的棕色烧瓶中,加入10ml的乙腈,在恒温油浴中,开启磁力搅拌,转速为200r/min;当温度升至78℃时缓慢滴加7mmol的n,n-二甲基乙酰胺二甲基缩醛至溶液中,在常压下反应15min;将反应液吸出移入分液漏斗中,冷却至室温,静置分层后取下层深棕色液体;在压力5kpa,温度50℃条件下旋蒸10min除去残留溶剂,即得到深棕色液体,即为产物n’-(4-十四烷苯基)-n,n-二甲基乙脒。
图1为产物n’-(4-十四烷苯基)-n,n-二甲基乙脒的1hnmr(cdcl3,500m)谱图,图2为产物n’-(4-十四烷苯基)-n,n-二甲基乙脒的红外谱图,谱图中3000-3200cm-1处有弱吸收峰,为苯环上碳氢键的吸收峰;1400-1600cm-1处四条谱带为苯环上的共轭键吸收峰;1600cm-1处的吸收峰为碳氮双键吸收峰。
核磁氢谱分析产物纯度为97%。目标产品总的收率为90%。
实施例2
本实施例提供了一种n’-(4-十四烷苯基)-n,n-二甲基甲脒,通过以下步骤合成得到:
将9mmol的4-十四烷基苯胺加至带有回流装置的棕色烧瓶中,加入15ml的乙腈,在恒温油浴中,开启磁力搅拌,转速为200r/min;加热,温度升至80℃时,缓慢滴加18mmol的n,n-二甲基甲酰胺二甲基缩醛至溶液中,在常压a条件下反应15min;将反应液吸出移入分液漏斗中,冷却至室温,待溶液静置分层后下,取下层深棕色液体;在压力5kpa,温度50℃条件下旋蒸10min除去残留溶剂,得到深棕色液体即为产物n’-(4-十四烷苯基)-n,n-二甲基甲脒。
用1hnmr(cdcl3,500m)分析产物,纯度为98%。目标产品总的收率为92%。
实施例3
本实施例提供了一种n’-(4-十二烷基苯基)-n,n-二甲基乙脒,通过以下步骤合成得到:
将5mmol的4-十二烷基苯胺加至带有回流装置的烧瓶中,加入11ml的乙腈,在恒温油浴中,开启磁力搅拌,转速为200r/min;加热,温度升至80℃时,缓慢滴加6mmol的n,n-二甲基乙酰胺二甲基缩醛至溶液中,在常压条件下反应15min;将反应液吸出移入分液漏斗中,冷却至室温,待溶液静置分层后,取下层深棕色液体;在压力10kpa,温度50℃条件下旋蒸10min除去残留溶剂,得到深棕色液体即为产物n’-(4-十二烷基苯基)-n,n-二甲基乙脒。
用1hnmr(cdcl3,500m)分析产物,纯度为96%。目标产品总的收率为90%。
实施例4
本实施例提供了一种n’-(4-十六烷苯基)-n,n-二甲基乙脒,通过以下步骤合成得到:
将5mmol的4-十六烷基苯胺至带有回流装置的烧瓶中,加入15ml的乙腈,在恒温油浴中,开启磁力搅拌,转速为200rad/min;加热,温度升至82℃时,缓慢滴加6mmol的n,n-二甲基乙酰胺二甲基缩醛至溶液中,在常压条件下反应20min;将反应液吸出移入分液漏斗中,冷却至室温,待溶液静置分层后,取下层深棕色液体;在压力10kpa,温度50℃条件下旋蒸10min除去残留溶剂,得到深棕色液体即为产物n’-(4-十六烷苯基)-n,n-二甲基乙脒。
用1hnmr(cdcl3,500m)分析产物,纯度为96%。目标产品总的收率为91%。
实施例5
本实施例提供了一种n’-(4-十八烷苯基)-n,n-二甲基乙脒,通过以下步骤合成得到:
将5mmol的4-十八烷基苯胺至带有回流装置的烧瓶中,加入20ml的乙腈,在恒温油浴中,开启磁力搅拌,转速为200r/min;加热,温度升至82℃时,缓慢滴加7mmol的n,n-二甲基乙酰胺二甲基缩醛至溶液中,在常压条件下反应20min;将反应液吸出移入分液漏斗中,冷却至室温,待溶液静置分层后,取下层深棕色液体;在压力10kpa,温度50℃条件下旋蒸15min除去残留溶剂,得到深棕色液体即为产物n’-(4-十八烷苯基)-n,n-二甲基乙脒。
用1hnmr(cdcl3,500m)分析产物,纯度为95%。目标产品总的收率为90%。
以上实施例说明,本发明的n’-(4-烷苯基)-n,n-二甲基脒的合成方法,n’-(4-烷苯基)-n,n-二甲基脒的收率可达90%,产品的纯度≧95%,反应操作简单,反应时间缩短,萃取剂乙腈易于回收。
显然,本发明的上述实施例仅是为清楚地说明本发明所作的举例,而并非是对本发明的实施方式的限定,对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动,这里无法对所有的实施方式予以穷举,凡是属于本发明的技术方案所引伸出的显而易见的变化或变动仍处于本发明的保护范围之列。
- 还没有人留言评论。精彩留言会获得点赞!