一种耐辐射奇球菌DNA甲基转移酶的制作方法
本发明涉及一种耐辐射奇球菌dna甲基转移酶m.drar1,涉及其所识别的dna保守序列特征及其甲基化修饰的模式。
背景技术:
dna修饰可引起染色质结构、dna构象及其稳定性变化,导致dna与蛋白质相互作用发生改变,调控基因表达的同时也扩展了dna的结构复杂性和信息深度。四种碱基的化学修饰传递的表观信息存在于包括病毒和噬菌体等在内的所有生命体中。dna甲基化修饰是一种广泛存在且非常重要的dna表观修饰现象,对生物体的生长、维持基因组稳定性以及分化等具有重要的调节作用。
介导催化dna甲基化修饰的酶主要是dna甲基化转移酶,根据形成产物不同,主要分为3类。其中有两类酶作用于碱基环外的氨基基团,产生m-6a和m-4c甲基化修饰。另外一种甲基化酶催化产生m-5c。在细菌不同种之间,甲基化酶的数量及其所识别的保守motif存在广泛的变异性,即使在亲缘关系较近的菌株中,酶的数量和识别位点也不相同。dna甲基转移酶已广泛应用于基因工程改造、分子生物学实验甚至是细菌感染的药物靶点等重要领域,开发利用新型的dna甲基转移酶对生命科学研究具有重大的意义。
耐辐射奇球菌具有极强的dna损伤修复能力,对由电离辐射、紫外线、干燥以及各种dna损伤化学试剂引起的突变和致死效应都具有超强的抗性。该细菌强悍的抗辐射能力得益于其多方面的特性,如具有一些不同于其它生物体的新型基因或新功能。其中体内m.drar1基因(基因名为dr_c0020,蛋白序列id为anc73351.1)表达的产物dna甲基转移酶m.drar1由434个氨基酸组成,具有典型的dna甲基转移酶保守结构域,属α型dna甲基转移酶类,但是,m.drar1的具体修饰模式及底物识别序列却未见报道。
技术实现要素:
为了克服现有技术的不足,本发明的目的是提供一种耐辐射奇球菌dna甲基转移酶。
一种耐辐射奇球菌dna甲基转移酶,从n端至c端依次为含fxgxg保守序列的adomet结合域、目标序列识别域及含tsppy保守序列的催化域,属于α型dna甲基转移酶类;所述的dna甲基转移酶的氨基酸序列如seqidno.1所示,或者如seqidno.1所示的氨基酸序列经过取代、缺失或者添加一个或者几个氨基酸且具有dna甲基转移酶活性的氨基酸序列;所述的dna甲基转移酶,它识别的dna保守序列为5’-ccgcgg-3’,并可甲基化修饰第二个胞嘧啶c上n4位,产生4mc类型的修饰碱基。
所述dna甲基转移酶反应缓冲液含50-200mmkcl、10-50mmtris-hclph7.5-8.0、0.1mmedta、3-7mmβ-me、20-100μmsam。
所述dna甲基转移酶的酶活性温度范围为4-60℃。
所述dna甲基转移酶的最适温度范围为25-37℃。
本发明的有益效果:
本发明提供了一种未被报道过的dna甲基转移酶及其识别的保守序列和甲基化修饰类型,可为新型限制-修饰酶的开发应用提供基础数据,对开发新型分子生物学的工具酶具有重要意义。
附图说明
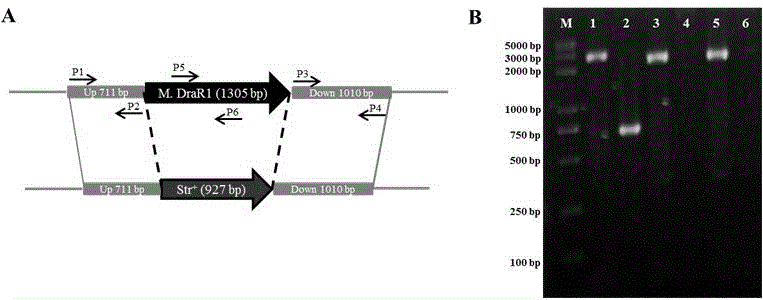
图1是本发明dna甲基转移酶m.drar1基因敲除株构建原理及pcr鉴定结果图。a部分为敲除原理示意图;b部分为pcr鉴定结果,其中,泳道1、3和5为引物p1p4扩增结果,泳道2、4和6为引物p5p6内部验证结果,泳道1和2为野生型drar1对照组,泳道3和4为m.drar1基因敲除株在ts培养基培养实验组,泳道5和6为m.drar1基因敲除株在tgy培养基培养实验组。
图2是耐辐射奇球菌野生型基因组dna经三代单分子测序分析4mc甲基化修饰“ccgcgg”motif的代表图,但m.drar1基因敲除株基因组同样的“ccgcgg”motif没有检测到4mc甲基化修饰,表明m.drar1所识别的保守motif为“ccgcgg”。
图3是耐辐射奇球菌野生型drar1和m.drar1基因敲除株基因组中4mc修饰的含量,用m4dc/dc的千分比显示。
图4是dna甲基转移酶m.drar1保守结构域示意图,m.drar1具有典型的保守结构域,从n端至c端依次为含“fxgxg”保守序列的adomet结合域、目标序列识别域(trd序列)及含“tsppy”保守序列的催化域,属α型n4-cytosinedna甲基转移酶类。其中,红底黑字代表保守氨基酸残基,粉底灰字为相对保守残基,浅蓝底白字为不保守残基。
图5是dna甲基转移酶m.drar1体内甲基化酶活作用分析示意图。其中,a部分为prrs-m.drar1质粒经限制性内切酶hindiii和sacii酶切位置示意图;b部分为prrs-m.drar1质粒及其pcr产物经hindiii和sacii酶切后琼脂糖电泳结果图,1和3泳道分别为prrs-m.drar1质粒pcr产物及其质粒本身未经酶切的效果,2和4则为经酶切后的结果图。
图6是dna甲基转移酶m.drar1体外甲基化酶活作用分析示意图。其中,泳道1为原始底物对照组,即没有用sacii进行切割;泳道2为未被甲基化阴性对照,即没有用目的蛋白甲基化修饰但用sacii进行了切割;泳道3、4和5分别为25℃、30℃和37℃反应条件下的甲基化修饰且sacii进行切割的阳性结果。
具体实施方式
以下结合附图和具体实施例对本发明做进一步的说明。
实施例1:耐辐射奇球菌dna甲基转移酶m.drar1底物识别特异性
(1)dna甲基转移酶m.drar1基因缺失株的构建:采用经典“三段链接法”将m.drar1基因序列的上游片段(约710bp)和下游片段(约1000bp)与抗性基因片段(链霉素抗性基因str,约927bp)通过t4连接酶(takara公司)体外链接成一个整体片段(见图1a),然后将这段三连产物转化耐辐射奇球菌感受态细胞,在含str抗性的tgy培养基平皿上筛选突变株,经pcr和测序验证得到m.drar1基因的缺失株—δm.drar1(见图1b)。
(2)耐辐射奇球菌野生型drar1和m.drar1基因敲除株基因组的提取:挑取野生型drar1和敲除株δm.drar1单菌落分别接种至5mltgy和ts(tgy含终浓度为10μg/ml的str抗生素)液体培养基,在28±5℃温度下,220rpm震荡培养35-40h后,各取50μl接种至50mltgy和ts液体培养基中,相同培养条件下培养至o.d.600为1.0-3.0之间,12000rpm离心收集菌体,根据细菌基因组dna提取试剂盒操作(天根生物)提取基因组dna。
(3)pacbio三代测序(single-moleculereal-timetechnology,smrt):经电泳检测合格的dna样本用covarisg-tube打断成构建文库所需大小的目的片段,经dna损伤修复及末端修复后,使用dna黏合酶将发卡型接头链接至dna片段两端,使用ampurepb磁珠对dna片段进行纯化选择,构建smrtbell文库。纯化后的片段经buffer回溶后,使用bluepipin片段筛选特定大小的片段,进一步磁珠纯化。构建好的文库经qubit浓度定量,并利用agilent2100检测插入片段大小,随后用pacbiorsii平台进行测序分析。结果表明所述dna甲基转移酶m.drar1可识别的dna保守序列为5’-ccgcgg-3’,并可甲基化修饰第二个胞嘧啶c上n4位,产生4mc类型的修饰碱基(见图2和表1)。表1是耐辐射奇球菌野生型drar1和m.drar1基因敲除株基因组中“ccgcgg”dna保守基序甲基化状态统计表。
(4)超高效液相三级四重杆质谱(uhplc-qqq-ms/ms)分析:2-4μg基因组dna溶于无核酸酶水,高温变性后即刻冰浴,先后用核酸酶p1、磷酸二酯酶i以及碱性磷酸酶进行充分酶解,酶解成单核苷后的样本稀释2倍后进行过滤,取若干微升样本进样分析。用标准曲线对不同的单核苷进行定量分析后计算修饰碱基与标准碱基的千分比值,结果说明所述m.drar1甲基化酶为n4-cytosinedna甲基转移酶(见图3)。
(5)dna甲基转移酶m.drar1生物信息学分析:采用clcsequenceviewer7软件将seqidno.1中的氨基酸序列与其它n4-cytosinedna甲基转移酶蛋白序列进行比对分析,结果进一步说明所述m.drar1甲基化酶为n4-cytosinedna甲基转移酶(见图4)。m.drar1甲基化酶可识别的dna保守序列为“ccgcgg”,与其它n4-cytosinedna甲基转移酶识别的保守序列均不相同,说明该基因编码的dna甲基转移酶为一种新型蛋白酶,可能贡献于耐辐射奇球菌特有的抗性机制(见表2)。表2是本发明所选用作生物信息学分析的n4-cytosinedna甲基转移酶氨基酸数目、识别保守序列及修饰位置、蛋白id以及物种信息统计图。
实施例2:dna甲基转移酶m.drar1体内酶活分析
(1)体内表达载体prrs-m.drar1载体的构建:以耐辐射奇球菌基因组dna为模板,采用pfu高保真聚合酶(全式基因,ap221)体外pcr扩增m.drar1基因,上游引物prrs-m.drar1-f含sbfi酶切位点和ttaagg框及ttaatcat序列,序列如seqidno.2所示;下游引物prrs-m.drar1-r含bamhi酶切位点和ccgcgg底物保守序列,序列如seqidno.3所示。1.0%琼脂糖电泳检测,纯化pcr产物(life公司dna纯化试剂盒,catno.116401),纯化后的pcr片段和prrs质粒(neb公司roberts教授馈赠)同时用sbfi和bamhi(neb公司)进行酶切,纯化酶切后的片段,用t4dna连接酶进行连接,连接产物转入大肠杆菌e.colier2796感受态细胞,方法参照《分子克隆指南》中的转化步骤。从la固体培养皿(la培养基中加入终浓度为100μg/ml氨苄青霉素amp)挑选克隆子进行测序鉴定(上游测序引物prrs-f序列如seqidno.4所示,下游测序引物prrs-r如seqidno.5所示),测序结果正确的即为阳性克隆。
(2)prrs-m.drar1质粒的提取:挑取阳性克隆子接种于5mlla液体培养基,37℃,220rpm震荡培养过夜,吸取50μl接种于50mlla液体培养基,相同培养条件下培养20±2h后,12000rpm离心收集菌体,采用质粒提取试剂盒提取质粒(dna美国axygen公司,cat.no.ap-mn-p-250g)。
(3)prrs-m.drar1质粒体外pcr扩增:采用pfu高保真聚合酶对prrs-m.drar1质粒进行体外扩增,上游扩增引物prrs-f如seqidno.4所示,下游扩增引物prrs-pcr-r如seqidno.6所示。扩增后的质粒经纯化后用作后续的阴性对照研究。
(4)m.drar1甲基转移酶活性分析:将以上获得的质粒用限制性内切酶hindiii和sacii进行酶切分析,因(2)中质粒“ccgcgg”序列被m.drar1甲基化酶甲基化,因此可避免其被sacii切割,而(3)中质粒没有被甲基化,所以会被切割,结果说明所述dna甲基转移酶m.drar1在体内可甲基化“ccgcgg”序列(见图5b)。
实施例3:dna甲基转移酶m.drar1体外酶活分析
(1)prrs-m.drar1质粒线性化处理:将以上获得质粒用hindiii切割成线性片段,纯化回收后备用。
(2)dna甲基转移酶m.drar1体外反应体系:将上述经过处理的质粒1-2μg加入含50-200mmkcl、10-50mmtris-hcl(ph7.5-8.0)、0.1mmedta、3-7mmβ-me及20-100μmsam的反应buffer中,另外加入1μm纯化后的m.drar1蛋白,置于适于条件下反应。
(3)dna甲基转移酶m.drar1的酶活最适酶活温度范围:将以上反应体系分别置于4-60℃反应0.5-1h,纯化反应结束后质粒dna或噬菌体dna,利用限制性内切酶sacii进行切割,结果进一步表明m.drar1甲基转移酶可甲基化修饰“ccgcgg”保守基序,其最适温度范围为25-37℃。在4℃时,也具有较弱的甲基化活性;在45-55℃温度范围之间仍具有甲基化活性,但活性很弱(见图6)。
本发明实施例中所用的菌株为耐辐射奇球菌(deinococcusradioduransr1,atcc13939),但根据本发明的教导和启示,任何人工合成或者其他自然含有的蛋白及其衍生物,如具有与dna甲基转移酶m.drar1的同源序列和类似的结构与功能,也在本发明的保护范围之内。
序列表
<110>浙江大学
<120>一种耐辐射奇球菌dna甲基转移酶
<160>6
<170>siposequencelisting1.0
<210>1
<211>434
<212>prt
<213>耐辐射奇球菌(deinococcusradiodurans)
<400>1
metthrglnproleuleupheaspleuprothrproargprothrtyr
151015
argaspthralaphealaserasnlysthrleualamethisargtrp
202530
valasntrpilealaglyphesersergluphevalglnhisalaleu
354045
gluleuhisleuproaspproasnprogluglnvalvalleuasppro
505560
pheglyglyvalglythrthrproilethralapheleuargglyhis
65707580
servalvalsertyraspileasnpropheproleuleuvalglnarg
859095
alalysleuargalaileglnaspvalthrproalagluphealagln
100105110
glnileglualaphethralahismetalathrglyglyvalprolys
115120125
serlysvalproglnglyphethrserargthrprophetyrserglu
130135140
lysvalleuvallysvalleuhisvaltrpasppheileasngluval
145150155160
alaaspgluaspleuargaspleupheglnvalalapheglyalathr
165170175
metvalsertyrserasntyrsertyrgluproserleuglyserarg
180185190
alaalaalaglylysproaspilegluaspalaaspvalalaglnval
195200205
metargasplysleuleuglumethisalaaspleuleuglyvalgln
210215220
glyilelysleuglyglyglnthralaglnvaltyrglnglyserphe
225230235240
metargsergluleuproaspserservalaspleumetvalthrser
245250255
proprotyrleuasnasntyrhistyrleuargasnthrargprohis
260265270
leutyrtrpleuglytyralathrserprolysaspleuargtyrleu
275280285
gluleuaspasntyrglylystyrtrpglnthrvalargaspalalys
290295300
tyrglnthrserleuilepheaspserprotrpleuglnaspleuval
305310315320
asnglnleualaglyvalglnseraspargglyvaltyrglyglygln
325330335
glytrpalaasntyralathrglutyrpheasnaspthrtyrargphe
340345350
leuglnlysthrglnalavalleuargproglyalalysalaleuile
355360365
valvalglyasnserilevallysglythrasnleuproileaspglu
370375380
valphethrhisilealaglnhisleuglypheserglyhisaspile
385390395400
hismetvalargaspserargileglyserserilevalglythrgly
405410415
leuargsergluglylysglyargleutyrglualavalvalgluleu
420425430
thrarg
<210>2
<211>48
<212>dna
<213>人工序列(artificialsequence)
<400>2
tttcctgcaggttaaggttaatcatatgacgcaacctcttctctttga48
<210>3
<211>55
<212>dna
<213>人工序列(artificialsequence)
<400>3
tttggatccccgcggttagtggtggtggtggtggtgcctggtcagttcaaccacg55
<210>4
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>4
accccaggctttacactttatgct24
<210>5
<211>23
<212>dna
<213>人工序列(artificialsequence)
<400>5
gcacagatgcgtaaggagaaaat23
<210>6
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>6
agcataaagtgtaaagcctggggt24
- 还没有人留言评论。精彩留言会获得点赞!