一种具有免疫抑制活性的PKSI型聚酮类化合物及其制备方法和应用与流程
本发明涉及一种具有免疫抑制活性的pksi型聚酮类化合物及其制备方法和应用,属于医药生物技术领域。
背景技术:
免疫抑制药物是指针对机体免疫应答有抑制作用的药物,是针对于自身免疫性疾病而研发出来的,能够抑制与免疫反应有关细胞的增殖和功能,能降低机体的免疫反应。临床上用于治疗自身免疫性疾病和移植排斥反应的免疫抑制药物主要有环孢菌素a(csa)、他克莫司(fk506)等,虽然此类药物在临床的疗效十分确切,但却具有不同程度的肝肾毒性或神经毒性等,长期服用会引发高脂血症、代谢性骨病甚至诱发肿瘤的发生;同时,这些免疫抑制药物的价格也比较昂贵。因此,急需寻找一种低毒、有效、价格低廉的免疫抑制药物,用于替代环孢菌素a(csa)、他克莫司(fk506)等。
技术实现要素:
本发明提供一种具有免疫抑制活性的pksi型聚酮类化合物及其制备方法和应用。从天然产物中筛选得到能抑制钙调蛋白磷酸酶活性的化合物,为自身免疫性疾病的治疗提供一种全新的治疗药物。
本发明采取的技术方案如下:
一种具有免疫抑制活性的pksi型聚酮类化合物,属于c2-不对称的十六元大环内二酯类,所述化合物命名为efophylinb,该化合物结构式(i)为:
本发明还提供了化合物efophylinb的制备方法,包括以下步骤:
s1、将链霉菌进行发酵培养;
s2、发酵培养结束后,用甲醇充分浸泡,过滤获取甲醇提取物,减压浓缩,回收甲醇,获得菌株发酵产物;
s3、取发酵产物,以乙酸乙酯、二氯甲烷、正己烷和甲醇的混合溶液为洗脱剂,进行硅胶洗脱,再依次进行c18反相柱梯度洗脱和葡聚糖凝胶柱洗脱,得到洗脱产物;
s4、取洗脱产物,选用c18反相柱,通过高效液相色谱法进行梯度洗脱,得到具有免疫抑制活性的pksi型聚酮类化合物efophylinb。
优选的,所述链霉菌为streptomycessp.dsm4137,保藏于中国典型培养物保藏中心(中国武汉.武汉大学),保藏日:2018年08月01日,保藏编号:cctccno:m2018512。
优选的,步骤s1中,发酵培养的条件为:将种子培养液接种到sfm固体发酵培养基,30℃培养箱中培养6d,获得菌株的发酵培养物。
优选的,步骤s2中,甲醇的浸泡时间为2h。
优选的,步骤s3为:取发酵产物,以乙酸乙酯、二氯甲烷、正己烷和甲醇的混合溶液为洗脱剂,按体积比9:6:6:8进行硅胶洗脱,每50ml为一个流份收集,tlc点板后合并类似流份,共获得10个组分,取组分5,经c18反相柱梯度洗脱,然后经sephadexlh-20葡聚糖凝胶柱洗脱,得到洗脱产物。
优选的,步骤s3中,c18反相柱的洗脱剂体积浓度70%~100%的甲醇水。
优选的,sephadexlh-20葡聚糖凝胶柱的洗脱剂为体积浓度80%的甲醇水。
优选的,步骤s4中,高效液相色谱的条件为:采用色谱柱agilenttechnologies1200,以甲醇-水为流动相进行梯度洗脱,流动相流速5ml/min,进样量25μl,梯度程序为:
本发明还进一步发现化合物efophylinb具有免疫抑制活性,可用于制备针对器官移植或自身免疫性疾病的免疫抑制药物。
与现有技术相比,本发明的有益效果是:
本发明涉及到化合物efophylinb,该化合物由链霉菌streptomycessp.dsm4137进行sfm固体培养基发酵并对发酵产物进行分离纯化获得。实验证明该化合物具有显著的钙调蛋白磷酸酶抑制活性,其免疫抑制作用比临床广泛使用的免疫抑制剂环孢菌素a低,且对脾细胞毒性较小,是一种高效、低毒的免疫抑制剂,可用于制备针对器官移植和自身免疫性疾病的第二代免疫抑制药物。
本发明化合物的cn酶抑制活性(ic50=24.04±0.37μm)远低于环孢菌素a(ic50=33.98±0.30μm),在药物浓度为56μm时,cn酶抑制率可达到67.44%;本发明化合物对正常小鼠的淋巴细胞有很弱的细胞毒性(ic50=48.98±1.25μm),而csa则严重影响淋巴细胞的存活率,具有很强的细胞毒性(ic50=10.15±0.42μm),可见efophylinb一定浓度范围内对淋巴细胞的毒性很低。
此外,本发明化合物来源于天然产物,且提取过程中未加入高毒性试剂,是一种低毒、有效、价格低廉的免疫抑制药物。
附图说明
图1:菌株streptomycessp.dsm4137的形态特征鉴定结果图。
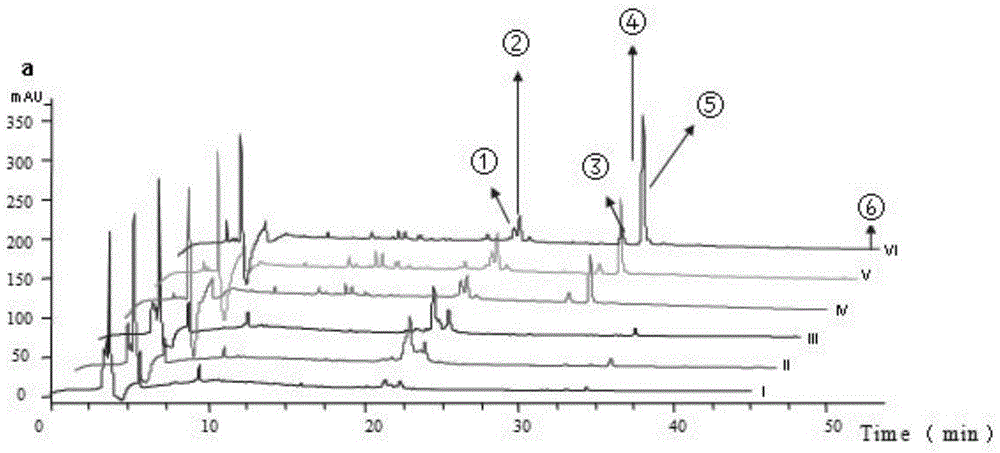
图2为固体发酵培养基培养结果图:tsby固体发酵培养基×2天(i);tsby固体发酵培养基×4天(ii);tsby固体发酵培养基×6天(iii);sfm固体发酵培养基×2天(iv);sfm固体发酵培养基×4天(v);sfm固体发酵培养基×6天(vi)。
图3为液体发酵培养基培养结果图:tsby液体发酵培养基×2天(i);tsby液体发酵培养基×4天(ii);tsby液体发酵培养基×6天(iii);sfm液体发酵培养基×2天(iv);sfm液体发酵培养基×4天(v);sfm液体发酵培养基×6天(vi)。
图4:化合物efophylinb的(+)hr-esi-ms质谱。
图5:化合物efophylinb的氢谱(500mhz,氘代氯仿)。
图6:化合物efophylinb的碳谱(500mhz,氘代氯仿)。
图7:化合物efophylinb的dept谱(500mhz,氘代氯仿)。
图8:化合物efophylinb的hsqc谱(500mhz,氘代氯仿)。
图9:化合物efophylinb的cosy谱(500mhz,氘代氯仿)。
图10:化合物efophylinb的hmbc谱(500mhz,氘代氯仿)。
图11:化合物efophylinb的roesy谱(500mhz,氘代氯仿)。
图12:化合物efophylinb的esi-ms/ms质谱。
图13:efophylinb和环孢菌素a的钙调蛋白磷酸酶靶酶活性。测试结果图。
图14:efophylinb和环孢菌素a的脾细胞毒性测试结果图。
具体实施方式
下面通过具体实施方式结合附图对本发明作进一步详细说明。
本发明实施例所用的实验方法如无特殊说明,均为常规方法。
本发明实施例所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
实施例一:streptomycessp.dsm4137的培养及鉴定
zeeck首次在希腊土壤样品中分离获得微生物样品streptomycessp.dsm3816,所述菌株streptomycessp.dsm4137是streptomycessp.dsm3816或其突变株,由英国剑桥大学提供,该菌株在sfm培养基中30℃恒温培养,形成广泛分枝的底物菌丝(直径为0.3-0.5μm)和气生菌丝,其分化成紧密的螺旋孢子链。孢子的外观范围从圆柱形到桶形(1.3-1.0×1.5pm),孢子表面具有皱纹。基质菌丝体的颜色倾向于棕色/灰色或黄色/灰色。成熟时,气生菌丝分化为紧密,螺旋状皱纹,圆柱形孢子链。形态鉴定结果见图1。
实施例二:streptomycessp.dsm4137的发酵条件优化
取菌种适量,接种到sfm固体平面培养基(大豆粉2%,d-甘露醇2%,琼脂2%)上,在30℃培养箱中培养6d。用接种针挑取适当孢子和菌体,接种到含有5ml的tsby种子培养液(胰蛋白大豆肉汤3%,蔗糖10.3%,酵母提取物0.5%)的25ml三角烧瓶中,置于摇床中,在30℃,150rpm条件下培养48h,获得种子培养液。
取该种子培养液(0.2ml),接种到tsby液体发酵培养基、sfm液体发酵培养基、tsby固体发酵培养基、sfm固体发酵培养基上,30℃培养箱中培养2d、4d、6d,获得菌株的发酵培养物,hplc-esi-ms采用5μc18柱子(4.6×250mm,phenomenex),表1中溶剂系统进行针对发酵培养基和培养时间的代谢产物筛选。结果见图2和图3。
表1hplc-esi-ms溶剂系统
通过图2和图3可知,实验确定发酵培养条件为:将种子培养液,接种到sfm固体发酵培养基,30℃培养箱中培养6d,获得菌株的发酵培养物。
图2中,序号①代表:
序号②代表:
序号③代表:
序号④代表:
序号⑤代表:
序号⑥代表:
实施例三、化合物efophylinb(i)的分离制备及结构鉴定
3.1菌株发酵产物提取
6l的sfm固体培养基发酵结束后,采用2倍体积甲醇充分浸泡2h,过滤获取甲醇提取物,减压浓缩,回收甲醇,重复三次,获得菌株streptomycessp.dsm4137发酵产物36g。
3.2薄层检测(tlc)快速辨识efophylinb
①点板
取少量发酵提取物用少量的溶剂溶解,用毛细管点于薄层硅胶gf254板上。点板的直径约2mm,距离薄层板约1.5厘米。点样后待其挥干,放入层析缸中。
②展开
展开剂加入层析缸后,将点样薄层硅胶板快速放入层析缸中,待溶剂前沿距离薄层硅胶板上端还有0.5cm时取出,晾干后显色。
③显色
将展开后的硅胶板,用2.0mol/lnaoh溶液喷涂显色后,即可看到桃红色等颜色反应,拍照记录显色结果。该方法适用于efophylinb的快速、早期、初步鉴别。
④溶剂系统的选择
对该菌发酵提取物薄层层析的展开系统进行摸索,最后决定用乙酸乙酯:二氯甲烷∶正己烷∶甲醇(9:6:6:8,v/v/v/v)洗脱。
3.3装柱及上样
吸附剂的填装,硅胶为多孔性物质,装柱宜用湿装法,发酵提取物为36g,将2.0kg硅胶混悬于二氯甲烷中,不断搅拌赶出气泡,连同溶剂一起倾入层析柱中。一次全部倾入,以免由于不同颗粒大小的硅胶沉降度不一样,使硅胶柱有明显的分段,容易影响分离效果,同时使硅胶柱层析表面保持在同一水平面有助于分离。将发酵提取物用少量的二氯甲烷溶解后,以1:1-1:2的量加入层析硅胶匀速研磨,将溶剂挥发至干呈松散状的样品,备用。将拌好的样品小心的铺在硅胶柱层析上,使样品表面保持平面,并在上面加上一层2-3cm的石英砂压出样品,有助于样品在硅胶柱层析中分离。在石英砂上面可以加入一些脱脂棉,起到缓冲的作用。
3.4efophylinb的制备
以乙酸乙酯:二氯甲烷∶正己烷∶甲醇(9:6:6:8,v/v/v/v)为洗脱剂,进行硅胶洗脱。每50ml为一个流份收集,tlc点板后合并类似流份,共获得10个组分;sfm-11(组分5)经c18反相柱70%~100%甲醇水梯度洗脱,初步得到纯化后组分sfm-11-2;sfm-11-2组分经sephadexlh-20葡聚糖凝胶柱80%甲醇水洗脱,得到组分sfm-11-2-2;制备柱色谱使用agilenttechnologies1200,以ch3oh和h2o为流动相梯度洗脱,流动相流速5ml/min,进样量25μl。梯度洗脱程序和dvd检测器的激发波长235nm(λ),按以下色谱条件进行制备,得到一定纯度的sfm-11-2-2-1,即efophylinb(2.8mg)。
表2hplc溶剂系统梯度表
3.5化合物的结构表征
运用现代各种光谱(ir,uv,cd)、波谱(1hnmr,13cnmr,dept,h-hcosy,hmqc,hmbc,noesy)和ms(esi-ms,hrms)等结构鉴定技术,确定所得到的活性成分化学结构,发现新颖结构活性成分。结果见图4-图11。
efophylinb(化合物i),白色固体(甲醇),阳离子hresi-ms(图4)在m/z913.5250处给出[m+na]+峰(calcdfor[m+na]+913.5289),因此该化合物分子式为c49h78o14,不饱和度为11,[α]20d+19.6(δc0.05,meoh)。经naoh溶液喷雾显色会呈现特征性的桃红颜色反应,uv254下呈紫色斑点,提示结构中存在共轭结构。1hnmr谱(图5)在低场区给出两组反式共轭二烯键信号,一组氢信号位于δh5.66(d,j=15.0hz,h-2),6.98(dd,j=15.0,11.1hz,h-3),6.09(dd,j=15.0,11.1hz,h-4)和5.62(dd,j=15.1,9.7hz,h-5);另一组氢信号位于δh5.64(d,j=15.0hz,h-2ˊ),6.95(dd,j=15.0,11.2hz,h-3ˊ),6.06(dd,j=15.0,11.2hz,h-4ˊ)和5.65(dd,j=15.0,9.1hz,h-5ˊ);在连氧区有1个甲氧基信号(δh3.05,s;δc46.6),在高场区有11个甲基(δh0.81-1.23)和23个sp3杂化的次甲基和亚甲基。综合分析13cnmr谱(图6)、dept(图7)和hsqc谱(图8)可以推断出该分子存在48个碳信号,包括:1个非共轭羰基δc203.1,2个酯羰基δc168.4和169.5,1个半缩酮碳δc99.0,10个烯次甲基,11个氧化次甲基(其中包括1个糖基异头碳δc103.2),1个甲氧基,8个脂肪族次甲基,3个亚甲基和11个甲基碳。将化合物i的1hnmr和13cnmr谱数据与已知物efophylin和elaiomycinm对比,发现三者非常相似,提示结构中含有相同的16元大环内二酯环,但化合物i的核磁信号相对更复杂,表明该化合物由两条不同的聚酮链聚合而成。结合1h-1hcosy谱以及hmbc谱给出的相关信号也证明了这一推测。1h-1hcosy谱(图9)提示该分子含有5个自旋系统。强hmbc相关(图10)从h2-12(δh2.31,dd,13.3,4.6)、h-15(δh3.48,m)、h3-19(δh1.01,d,7.0)到c-11(δc103.2),从h-22(5.03,brs)到c-13(δc70.1)、c-24(δc65.8)和c-26(δc66.1),揭示了efophylin的线性聚酮链;从h2-12ˊ(δh6.25,d,15.8)、h-15ˊ(δh3.81,m)、h3-19ˊ(δh1.17,d,7.2)到c-11ˊ(δc203.1)的hmbc相关,表明了efophylinm的线性聚酮链;由h-7(δh4.73,brd,10.2)到c-1ˊ(δc169.5)、h-7ˊ(δh5.06,brd,10.5)到c-1(δc168.4)的hmbc相关可推导出上述2条线性聚酮链通过c-1/c-7ˊ和c-1ˊ/c-7之间的两个内酯桥形成c2-不对称大环内二酯结构;另外,hmbc显示甲氧基11-och3(δh3.06,s)与c-11(δc103.2)相关,说明och3分别连接在大环内二酯母核c-11的位置上。由此化合物i的平面结构得以确定。以上结构可由esi质谱获得进一步确认,准分子离子m/z913[m+na]+的碰撞在m/z743和425产生碎片离子峰。该化合物的相对立体结构是通过化合物i的耦合常数、roesy相关(图11)与efophylin和elaiomycinm的对比来确定的;绝对构型与所有天然的efophylin相同,可通过构建聚酮化合物链的聚酮合酶的生物合成分析进行确认。该新化合物命名为efophylinb。其核磁数据如下:
表3.efophylinb在cdcl3中的1hnmr(500mhz)data(jinhz)和13cnmr(125mhz)data(jinhz)数据
经以上分析,确定efophylinb的结构式为:
实施例四efophylinb的钙调蛋白磷酸酶靶酶活性测试
将cna稀释成合适的酶溶液备用,取5ml试管若干置于冰上,分别依次加入10μl药和10μl酶液于冰上孵育5min,加入180μl测活液于30℃的水浴反应20min,加入1800μl终止液终止反应,720型分光光度计上测定od410值。空白对照:10μl酶稀释液+10μlbuffer;酶:10μl酶+10μlbuffer;药对照:10μl酶稀释液+10μl药;酶+药:10μl酶+10μl药(注:buffer为溶解药所用的溶液),按公式计算化合物i对cna的相对抑制率:
相对抑制率(%)=[1-(od药+酶-od药对照)/od酶]×100%
测试结果见表4和图13。
表4
实施例四小鼠脾淋巴细胞毒活性测试
将balb/c小鼠摘眼球放血断颈处死菌取脾,用rpmi1640培养液制成细胞悬液(密度为1.5×107个/ml)。然后将100μl细胞悬液、100μl的各浓度的药物加入96孔板;对照组加100μl含15%血清和0.1%dmso的培养液,于37℃,5%二氧化碳条件下培养68小时。efophylinb和csa的终浓度分别为1、5、10、15、20、30和40μm,dmso的终浓度最高为0.1%。等培养结束时,每孔加入20μlcck-8,继续放入培养箱中孵育。4小时后,酶标仪读取od450。结果见图14。脾细胞存活率=od实验组/od对照组×100%。
结果见表5和图14
表5
以上内容是结合具体的实施方式对本发明所作的进一步详细说明,不能认定本发明的具体实施只局限于这些说明。对于本发明所属技术领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干简单推演或替换。
- 还没有人留言评论。精彩留言会获得点赞!