一种具有AIE效应刚性共轭大环化合物及其制备和应用的制作方法
本发明属于刚性共轭大环化合物及其制备和应用领域,特别涉及一种具有aie效应刚性共轭大环化合物及其制备方法和应用。
背景技术:
自上世纪80年代以来,超分子化学经历了迅猛地发展。其中,开发拥有与常规硅等无机半导体材料不同特性的光电器件的研究正飞速发展,尤其是利用π-电子共轭体系的有机单分子、高分子为基本组成单元的材料,其设计和开发越来越活跃。与此相关联,拥有规整结构、π-共轭体系的刚性大环化合物及其衍生物正受到巨大的瞩目。这一类富π共轭化合物根据其极性官能团的位置和取向,通过本身所拥有的极强的π-π相互堆积作用很容易发生自组装,从而使大环形成固态一维柱状非共价键高分子聚集体结构,固态二维网状结构和三维纳米结构。这种高度有序的一维、二维、三维结构可为空穴、电子等载体提供可能的移动通道。作为崭新的显示各向异性特性的电子及光电子材料素材而引起了科学家们的极大的兴趣。
含n杂原子的共轭大环化合物作为一类新兴的共轭大环化合物由于其潜在的功能和应用受到广泛关注。含嘧啶基团的共轭大环化合物是此类化合物的代表之一,它们具有非常规整的形状结构,环直径处于纳米级别,根据其刚性环骨架和环上柔性链的不同,这类共轭大环化合物可通过自身π-π堆砌或者concave-convex相互作用自组装构筑一维、二维和三维超分子结构,也可以并用嘧啶所含n杂原子具有结合质子,金属离子等能力,用于制备复合材料研究以及能够感应外界刺激的响应性功能材料。因此,这类大环化合物作为优良的自组装单元分子应用到新型功能材料开发中。2015年,xiao等(xiaod,zhangd,chenb,etal.size-selectiverecognitionbyatubularassemblyofphenylene-pyrimidinylenealternatedmacrocyclethroughhydrogen-bondinginteractions.langmuir,2015,31(39):10649-55.)通suzuki偶联合成了具有嘧啶基团的刚性共轭大环,大环化合物可在二氯甲烷中组装成纳米管结构,可通过酸碱的调节,实现纳米管的可逆解离和重组装,有望进一步开发成为外界刺激响应性功能性超分子材料。
传统上的荧光物质大多在溶液中进行研究,在溶液中可以发出很强的荧光,但是随着溶液浓度的增加,荧光逐步发生淬灭。这是因为,在高浓度时共轭体系分子聚集出现π-π堆积,基于分子间的相互作用,增加了费辐射能的损耗,使激发态的能量逐渐衰减,出现发光效率低甚至淬灭,这种现象称为聚集淬灭现象(aggregationcausedquenching,acq)。2001年,tang等(jingdongluo,zhiliangxie,jackyw.y.lam,etal.aggregation-inducedemissionof1-methyl-1,2,3,4,5-pentaphenylsilole.chem.commun.,2001,1740-1741.)在研究六苯基噻咯分子(hps)中发现,在完全溶解时没有出现荧光,而加入不良溶剂水时,溶液的荧光逐渐增强,而且荧光强度随着含水量的增加而增加。经过研究发现,这是因为在不良溶剂水加入时,hps分子会发生聚集而逐渐形成聚集体,这一现象为聚集诱导发光(aggregationinducedemission,aie)。经过大量实验验证和理论模拟发现,这主要是由于在溶解的状态下,hps分子内的芳香环相对于中心共轭基元(芳香环或双键)活跃的运动耗散分子激发态能量,使得辐射衰变程度降低,出现荧光发射效率下降的现象;当不良溶剂加入后,这些分子发生聚集,分子内运动受限,辐射增强,出现强荧光现象。
自2001年唐本忠院士提出聚集诱导发光(aie)现象(jingdongluo,zhiliangxie,jackyw.y.lam,etal.aggregation-inducedemissionof1-methyl-1,2,3,4,5-pentaphenylsilole.chem.commun.,2001,1740-1741.)以来,受到了越来越多的科研工作者的关注,各种各样的aie化合物应运而生。其中四苯乙烯由于合成简单,性质稳定所以是研究最多的一类aie分子。在aie过程中,化合物在溶解状态是没有荧光的,在加入不良溶剂聚集时,化合物会出现较强的荧光。四苯乙烯分子是c2对称,四元反应位点可使其成为一种多功能的建筑模块用来合成有机配体、单体、聚合物等;四苯乙烯分子呈现独特的螺旋桨式分布,在聚集分子间不能紧密堆积,形成空隙,利于分子自组装;四苯乙烯分子在聚集态独特的发光现象和电子性质为oleds和荧光探针的设计提供了新思路。
技术实现要素:
本发明所要解决的技术问题是提供一种具有aie效应刚性共轭大环化合物及其制备方法,以克服现有技术中的嘧啶刚性大环化合物在荧光发光方面能力较弱,成环率较低的弊端。
本发明的一种具有aie效应刚性共轭大环化合物,所述化合物的结构式为:
式中,r为氢、疏水性基团、亲水性基团或手性基团。
所述r包括酯基、氰基、氨基、巯基中的一种或几种。
所述化合物的结构式为:
本发明的一种具有aie效应刚性共轭大环化合物的制备方法,包括:
(1)在惰性气氛下,将3,5-二溴苯酚、长链卤代烃或磺化有机物和第一碱以摩尔比为1.8-2.2:1:8-15与第一溶剂混合,进行亲核取代反应,得到3,5-二溴苯基长链醚;在惰性气氛下,将3,5-二溴苯基长链醚、双频哪醇合二硼、过渡金属催化剂和第二碱以摩尔比为1:2.1-2.5:0.12-0.18:5-8与第二溶剂混合,进行miyaura反应,得到3,5-二频哪醇硼酯苯基长链醚,其中3,5-二溴苯酚与第一溶剂的比例为3-4mmol:45-55ml;双频哪醇合二硼与第二溶剂的比例为0.6-0.7mmol:15-25ml;
(2)在惰性气氛下,将步骤(1)中3,5-二频哪醇硼酯苯基长链醚、5-溴-2-碘嘧啶、过渡金属催化剂和碱以摩尔比为1:2-4:0.05-0.15:8-12与溶剂混合,进行suzuki偶联反应,得到3,5-双(5-溴嘧啶)基苯基长链醚或3,5-双(5-溴嘧啶)基苯酚;其中5-溴-2-碘嘧啶与溶剂的比例为0.5-0.6mmol:12-20ml;
(3)在惰性气氛下,将二苯甲烷与n-buli搅拌反应,加入1,4-二溴苯甲酮,继续搅拌反应,淬灭反应,萃取,旋干,将旋干后的产物与对甲苯磺酸反应,得到1,1-二苯基-2,2-二(4-溴苯基)乙烯,在惰性气氛下,将1,1-二苯基-2,2-二(4-溴苯基)乙烯、pdcl2(pph3)2、cui和碱以摩尔比为1:0.05-0.15:0.05-0.15:40-45与溶剂混合,然后加入2-甲基3-丁炔-2-醇,进行sonogashira偶联反应,得到1,1-二苯基-2-(4-溴苯基)-2-(2-甲基-2-羟基-丁炔基)乙烯,其中二苯甲烷、n-buli、1,4-二溴苯甲酮与对甲苯磺酸的摩尔比为1-1.5:1-1.5:1:0.11-0.15;1,1-二苯基-2,2-二(4-溴苯基)乙烯与2-甲基3-丁炔-2-醇的摩尔比为1:1-1.3,1,1-二苯基-2,2-二(4-溴苯基)乙烯与溶剂的比例为3-4mmol:35-45ml;
(4)在惰性气氛下,将步骤(3)中1,1-二苯基-2-(4-溴苯基)-2-(2-甲基-2-羟基-丁炔基)乙烯、第一过渡金属催化剂、双频哪醇合二硼和第一碱以摩尔比为1:0.05-0.15:1.8-2.2:2.8-3.2与第一溶剂混合,进行suzuki偶联反应,得到1,1-二苯基-2-(4-频哪醇硼酯苯基)-2-(2-甲基-2-羟基-丁炔基)乙烯,在惰性气氛下,将1,1-二苯基-2-(4-频哪醇硼酯苯基)-2-(2-甲基-2-羟基-丁炔基)乙烯、步骤(2)中3,5-双(5-溴嘧啶)基苯基长链醚、第二过渡金属催化剂和第二碱以摩尔比为2.1-2.5:1:0.16-0.25:8-15与第二溶剂混合,再次suzuki偶联反应,得到化合物3,其中双频哪醇合二硼与第一溶剂的比例为0.3-0.5mmol:15-25ml;化合物3的结构式为:
(5)在惰性气氛下,将步骤(4)中化合物3与强碱以摩尔比为1:180-250与溶剂混合,脱去保护基,提纯,得到化合物2,在惰性气氛下,将化合物2、cucl与cucl2以摩尔比为1:50-60:8-15与无水吡啶混合,对接成环反应,提纯,得到刚性共轭大环化合物,其中化合物2的结构式为:
所述步骤(1)中第一碱为碳酸钾;第一溶剂和第二溶剂均为无水dmf。
所述步骤(1)中亲核取代反应温度为95-110℃,时间为48-55h。
所述步骤(1)中过渡金属催化剂为pd(dppf)cl2;第二碱为koac。
所述步骤(1)中miyaura反应温度为75-90℃,时间为2.5h-3.5h。
所述步骤(2)中碱为碳酸钾;过渡金属催化剂为pd(pph3)4;溶剂为thf和甲苯的混合溶剂。
所述步骤(2)中suzuki偶联反应温度为75-100℃,时间为40-50h。
所述步骤(3)中搅拌反应温度为-5-5℃,时间为30-60min。
所述步骤(3)中继续搅拌反应为:-5-5℃搅拌2-4h,升至室温反应过夜。
所述步骤(3)中与对甲苯磺酸反应温度为100-120℃,时间为12-24h。
所述步骤(3)碱为三乙胺;溶剂为thf;sonogashira偶联反应温度为75-90℃,时间为24-30h。
所述步骤(4)中第一过渡金属催化剂为pd(dppf)cl2;第一碱为koac;第一溶剂为dmf。
所述步骤(4)suzuki偶联反应温度为75-90℃,时间为12-15h。
所述步骤(4)第二过渡金属催化剂为pd(pph3)4;第二碱为碳酸钠;第二溶剂为1,4-二氧六环。
所述步骤(4)再次suzuki偶联反应温度为100-120℃,时间为48-60h。
所述步骤(5)中强碱为氢氧化钠;溶剂为甲苯。
所述步骤(5)中脱去保护基温度为110-130℃,时间为12-16h。
所述步骤(5)中对接成环反应温度为55-70℃,时间为5-8d。
本发明的一种具有aie效应刚性共轭大环化合物的应用。
所述刚性共轭大环化合物嘧啶基团上的n原子与pt、pd或ru配位。
所述刚性共轭大环化合物嘧啶基团上的n原子与pt配位后的结构式为:
所述刚性共轭大环化合物与金属配位后用于功能材料开发。
所述刚性共轭大环化合物作为自组装单元分子应用于功能材料开发。
所述刚性共轭大环化合物对ph的响应性具有可逆性,用于可调控荧光材料的研究。
所述刚性共轭大环化合物通过对客体分子的尺寸选择性感应制备具有不同光学及电学的复合材料。
所述刚性共轭大环化合物含四苯乙烯基团具有aie效应,可应用于新型荧光材料的研究。
所述刚性共轭大环化合物对甲苯有很好的响应性,甲苯可调控溶液的荧光光谱变化,可应用于甲苯的定性检测中。
本发明提供了一种含嘧啶基团刚性共轭大环化合物经自组装形成超分子结构体用于功能材料开发的研究。本发明的化合物具有规整的分子结构,孔径大小处于纳米级别,可通过自身π-π堆砌或者concave-convex相互作用自组装构筑一维、二维和三维超分子结构,作为优良的自组装单元分子应用到新型功能材料开发中;另外,还可以利用嘧啶所含n杂原子具有结合质子、金属离子等能力,用于制备复合材料研究以及能够感应外界刺激的响应性功能材料。
现有技术中的嘧啶刚性大环化合物,在荧光发光方面能力较弱,本发明为提高嘧啶大环化合物的荧光特性,在现有的嘧啶刚性大环化合物中引入四苯乙烯基团,制备具有ai效应的嘧啶刚性大环化合物;另一方面,现有技术(xiaod,zhangd,chenb,etal.size-selectiverecognitionbyatubularassemblyofphenylene-pyrimidinylenealternatedmacrocyclethroughhydrogen-bondinginteractions.langmuir2015,31(39):10649-55)中,大环的成环率较低,只有10%左右,而本发明将成环率提高到50%。
在新提出的甲苯调控光谱变化这一性能,这也是本专利首次发现并提出的理论,具有独特性、创新性。
有益效果
(1)本发明提供了一系列新型含嘧啶基团刚性共轭大环化合物的成环合成法,具备原料合成方法简单,制备工艺成熟,产率较高等优点;
(2)本发明刚性共轭大环化合物可作为优良的自组装单元分子应用到新型功能材料开发中;
(3)本发明刚性共轭大环化合物可通过自身所含的n杂原子与金属配位,呈现特殊的光电性能,用于复合材料研究;
(4)本发明刚性共轭大环化合物可通过对酸的感应制备可调控性荧光材料和电子活性材料(电子转移或能量转移);
(5)本发明刚性共轭大环化合物可通过对客体分子的尺寸选择性感应制备具有不同光学及电学的复合材料;
(6)本发明刚性共轭大环化合物含四苯乙烯基团,具有aie效应,可应用于新型荧光材料的研究;
(7)本发明刚性共轭大环化合物对甲苯具有很好的响应性,甲苯可调控荧光光谱的变化,有望应用于甲苯的定性检测。
附图说明
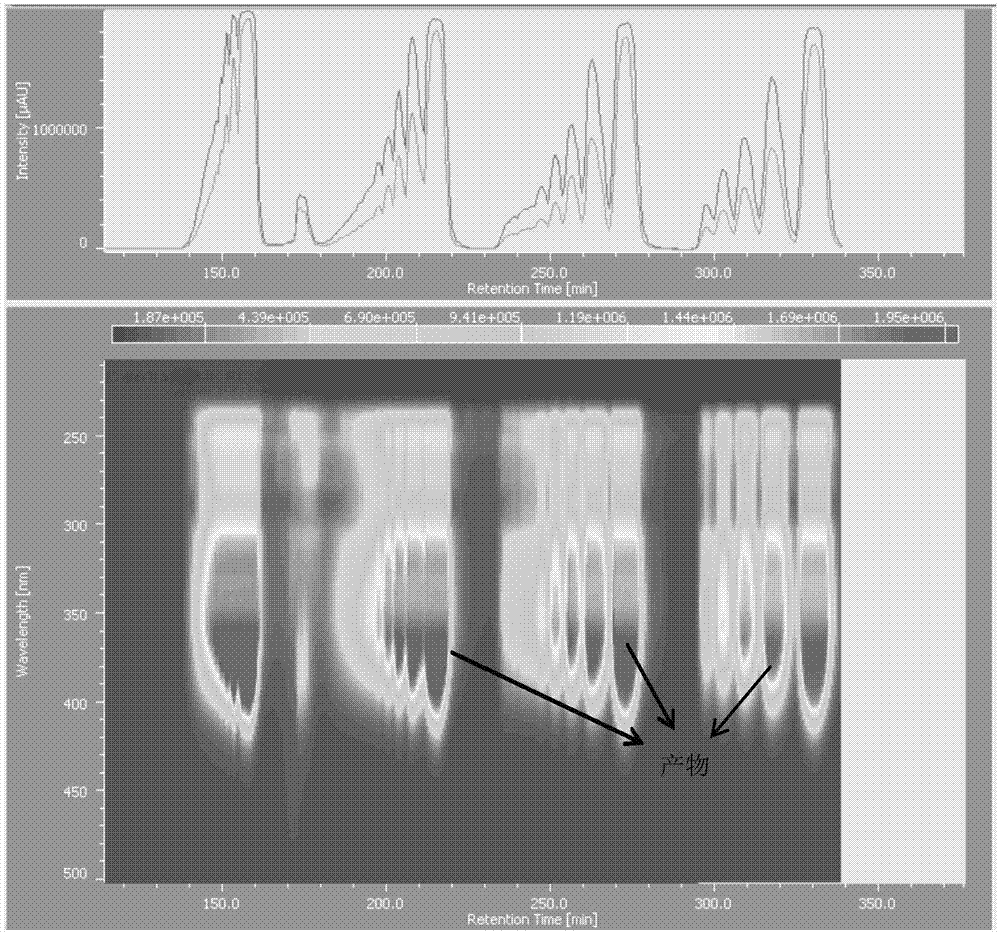
图1为实施例11中产物的gpc分离图;
图2为实施例11中目标产物的maldi-tofmass图;
图3(a)为实施例11中的产物在不同体积比例h2o/thf下的荧光光谱图;
图3(b)为实施例11中的产物在便携式手提紫外荧光灯(365nm)下不同体积比例h2o/thf中的荧光光谱图,其中图中从左到右各瓶中为含水比例(vh2o:vthf)分别为0、10%、20%、30%、40%、50%、60%、70%、80%、90%、95%的混合溶液或悬浊液;
图4(a)为实施例11中的产物对不同用量(0.025微升~7.0微升)芳香族化合物甲苯有一定的响应性;
图4(b)、(c)、(d)为图4(a)的光谱变化分解图,其中图4(b)是对0.025微升~0.175微升的甲苯,图4(c)是对0.200微升~0.400微升的甲苯,图4(d)是对0.425微升~7.000微升的甲苯。
具体实施方式
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。此外应理解,在阅读了本发明讲授的内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
所用化学试剂除pd(pph3)4、超纯水外,其它均购自aldrich、tci、百灵威或国药集团有限公司等。
所使用的溶剂和试剂除特殊说明外,其纯化参考perrin,d.d.;armarego,w.l.f.;perrin,d.r.inpurificationoflaboratorychemicals,pergamon:oxford,1980.
所使用柱层析固定相硅胶为300-400目(青岛)。
实施例以十二醇为原料,经取代、硼氢化氧化、磺化得到化合物4c,在碱性条件下与3,5-二溴苯酚通过亲核取代反应合成化合物4b,再与双频哪醇合二硼进行miyaura反应合成化合物4a,与5-溴-2-碘嘧啶通过suzuki偶联反应合成单体4;以1,4-二溴苯甲酮为起始原料,经过取代反应、sonogashira偶联反应、suzuki偶联反应合成单体5;利用单体5和单体4通过suzuki偶联合成含半环化合物3;半环化合物在强碱的作用下脱去保护基得到末端炔的半环化合物2,最后在催化剂的作用下,半环化合物对接成环,得到最终大环化合物1。反应方程式如下所示:
实施例1
化合物(4e)的合成:
在500ml的斜两口瓶中加入nah(8.4g,0.352mol,4.4eq.),在氩气氛围下依次加入250ml新蒸无水thf和1-十二醇(31g,0.167mol,2.1eq.),常温下搅拌,再加入3-氯-2-氯甲基丙烯(10g,0.08mol,1eq.)。最后温度升高至65℃下反应过夜,待反应液冷却至室温,缓慢的加入乙醇进行淬灭反应,旋蒸旋去thf,反应液用ch2cl2萃取,水洗,无水硫酸镁干燥,过滤,浓缩,粗产物用硅胶层析柱提纯(洗脱剂:pe/ch2cl2=5/1),得无色液体25g,产率75%。1hnmr(400mhz,cdcl3):δ(ppm)5.14(s,2h),3.95(s,4h),3.39(t,j=6.6hz,4h),1.62–1.48(m,4h),1.25(s,36h),0.87(t,j=6.7hz,6h).
实施例2
化合物(4d)的合成
在100ml的斜两口瓶中加入4e(5.0g,11.78mmol),抽换氩气三次后,在氩气氛围下加入80ml新蒸无水thf,在0℃下慢慢加入thf溶解的bh3(1mol/l,28ml),再快速搅拌3小时后加入18mlnaoh(3mol/l)溶液。再剧烈搅拌15分钟,滴加18mlh2o2(30%),最后室温下搅拌过夜后,加入k2co3至饱和,反应液经ch2cl2萃取,水洗,无水硫酸镁干燥,过滤,浓缩,粗产物用硅胶柱层析提纯,得无色液体3.5g,产率79%。1hnmr(400mhz,cdcl3):δ(ppm)3.78(d,j=5.1,2.4hz,1h),δ3.76(d,j=5.0hz,1h),δ3.68(d,j=6.1hz,1h),3.62–3.58(m,2h),3.52(m,j=9.4,6.0hz,2h),3.47–3.25(m,4h),2.17(s,1h),1.55(d,j=10.2,3.9hz,4h),1.46–1.03(m,36h),0.99–0.74(m,6h).
实施例3
化合物(4c)的合成
在100ml的斜两口瓶中加入4d(2g,4.5mmol,1eq.),tscl(3.91g,22.66mmol,5eq.),抽换氩气三次后,在氩气氛围下加入60ml无水ch2cl2,吡啶(5.3g,67.5mmol,15eq.),在25℃下快速搅拌5小时,反应液经ch2cl2萃取,合并有机相,有机相先用1mol/lhcl洗,再用蒸馏水洗,无水硫酸镁干燥,过滤,浓缩,粗产物用硅胶层析柱提纯(洗脱剂:ch2cl2),得淡黄色液体2.2g,产率85%。1hnmr(400mhz,cdcl3):δ(ppm)7.81(d,j=8.3hz,2h),7.36(d,j=8.0hz,2h),4.12(d,j=5.6hz,2h),3.46–3.23(m,8h),2.47(s,3h),2.20(q,j=11.5,5.8hz,1h),1.54–1.40(m,4h),1.37–1.17(m,36h),0.90(t,j=6.8hz,6h).
实施例4
化合物(4b)的合成
在100ml的斜两口瓶中依次加入3,5-二溴苯酚(838mg,3.35mmol,2eq.)4c(1g,1.677mmol,1eq.)和k2co3(2.3g,16.7mmol,10eq.),在氩气氛围下加入50ml无水dmf,升高温度至100℃下反应48h后停止反应。旋蒸除去dmf,经ch2cl2萃取,无水硫酸镁干燥,过滤,浓缩,粗产物用硅胶柱层析提纯(洗脱剂:pe/ea=25/1),得浅黄色液体0.95g,产率85%。1hnmr(400mhz,cdcl3):δ(ppm)7.25(t,j=1.5hz,1h),7.04(d,j=1.5hz,2h),4.02(t,j=10.2hz,2h),3.57–3.47(m,4h),3.42(t,j=6.6hz,4h),2.41–2.28(m,1h),1.56(d,j=13.5,6.7hz,4h),1.40–1.21(m,36h),0.88(t,j=22.0,7.4hz,6h).
实施例5
化合物(4a)的合成
在100ml的斜两口瓶中依次加入4b(200mg,0.295mmol,1eq.),pdcl2(dppf)(45.5mg,0.045mmol),koac(175mg,1.28mmol,6eq.)和双频哪醇合二硼(165mg,0.65mmol,2.2eq.),抽换氩气三次后,在氩气氛围下加入20ml无水dmf,升高温度至80℃下反应3小时,停止反应。待反应液冷却至室温后,经ch2cl2萃取,无水硫酸镁干燥,过滤,浓缩,粗产物用硅胶层析柱提纯(洗脱剂:pe/ea=20/1),gpc分离,得浅黄色油状物:230mg,产率51%。1hnmr(400mhz,cdcl3):δ(ppm)7.87(s,1h),7.46(s,2h),4.08(d,j=5.5hz,2h),3.56(d,j=6.0hz,4h),3.42(t,j=6.6hz,4h),2.37(m,j=11.5,5.7hz,1h),1.56(q,j=13.1,6.4hz,4h),1.36(s,24h),1.27(s,36h),0.90(t,j=6.7hz,6h).
实施例6
化合物(4)的合成
在25ml的斜两口瓶中依次加入4a(150mg,0.195mmol,1eq.),5-溴-2-碘嘧啶(166mg,0.585mmol,3eq.),抽换氩气三次后氩气保护下加入pd(pph3)4(22mg,0.019mmol,10%mol),氩气氛围下继续加入thf(冷冻除氧)/toluene(冷冻除氧)(10ml/5ml=2/1),2mol/lk2co3(冷冻除氧,0.98ml,10eq.),升高温度至80℃下回流,反应40小时后停止反应。待反应液冷却至室温后,旋蒸除去溶剂,经ch2cl2萃取,无水硫酸镁干燥,过滤,浓缩,粗产物经硅胶层析柱提纯(洗脱剂:pe/ea=20/1),得白色固体90mg,产率56%。1hnmr(400mhz,cdcl3):δ(ppm)9.08(s,1h),8.88(s,4h),8.15(d,j=1.4hz,2h),4.25(d,j=5.6hz,2h),3.62(d,j=6.0hz,4h),3.45(t,j=6.6hz,4h),2.54–2.38(m,1h),1.59(m,j=14.6,7.5hz,4h),1.24(s,36h),0.89(s,6h).
实施例7
化合物(7)的合成
在250ml的斜两口瓶中加入二苯甲烷(3.36g,20mmol,1.25eq),在氩气氛围下加入80ml新蒸的无水thf,0℃下搅拌30min。缓慢加入n-buli(1.6m,12.5ml,1.25eq),继续搅拌1h,然后加入1,4-二溴苯甲酮(5.44g,16mmol,1eq),继续反应2h后升至室温,反应过夜。加入氯化铵饱和水溶液淬灭反应,用dcm萃取三次,旋除溶剂,粗产物无需进一步提纯,直接用于下一步反应。在反应瓶中加入对甲苯磺酸(0.34g,1.8mmol),甲苯80ml,110℃回流过夜,冷却至室温,旋除甲苯,用dcm萃取三次,无水硫酸钠进行干燥,过滤,旋干。粗产物经硅胶层析柱提纯(洗脱剂:pe),得白色固体6.6g,产率75%。1hnmr(400mhz,cdcl3)δ7.25(d,j=8.1hz,4h),7.15(dd,j=9.2,5.8hz,6h),7.02(dd,j=5.9,2.5hz,4h),6.89(d,j=8.1hz,4h).
实施例8
化合物(6)的合成
在100ml两口瓶中加入化合物7(1.6g,3.265mmol,1eq),pdcl2(pph3)2(0.227g,0.3265mmol),cui(0.045g,0.3265mmol),氩气氛围下加入冷冻除氧的三乙胺20ml,冷冻除氧的thf40ml,固体溶解后加入2-甲基-3-丁炔-2-醇(0.329g,3.918mmol,1.2eq),80℃回流24h。冷却室温后,旋除溶剂,dcm萃取三次,用无水硫酸钠干燥并过滤,浓缩。粗产物经硅胶层析柱提纯(洗脱剂:dcm:pe=1:1),得淡黄色固体0.78g,产率48%。1hnmr(400mhz,cdcl3)δ7.25–7.20(m,2h),7.20–7.09(m,8h),7.02(d,j=2.2hz,4h),6.96(d,j=8.1hz,2h),6.88(d,j=8.4hz,2h),1.62(d,j=14.3hz,6h).13cnmr(101mhz,cdcl3)δ143.45(s),143.15(d,j=3.6hz),142.33(s),139.00(d,j=6.7hz),133.04(s),131.11(dt,j=26.9,11.5hz),127.89(t,j=9.0hz),126.89(t,j=4.7hz),120.87(s),120.73(s),94.25(s),82.18(s),77.44(s),77.12(s),76.80(s),65.63(s),31.53(s).maldi-tofmass:calcd.forc31h25bro[m]+:m/z=492.1083;found:492.0991.
实施例9
化合物(5)的合成
在50ml干燥的两口瓶中加入化合物6(100mg,0.2mmol,1eq),pd(dppf)cl2(22mg,0.03mmol,15%),双频哪醇合二硼(103mg,0.4mmol,2eq),醋酸钾(58.8mg,0.6mmol,3eq),冷冻除氧的dmf20ml,80℃下加热12h。冷却室温后,旋除溶剂,dcm萃取三次,用无水硫酸钠干燥并过滤,浓缩。粗产物经硅胶层析柱提纯(洗脱剂:dcm:pe=1:1),得淡黄色固体32mg,产率29.6%。1hnmr(400mhz,cdcl3)δ7.54(d,j=7.8hz,2h),7.15–7.06(m,8h),7.00(d,j=7.5hz,6h),6.93(d,j=8.1hz,2h),1.58(s,6h),1.31(s,12h).13cnmr(101mhz,cdcl3)δ146.32(s),143.84(s),143.39(s),143.27(s),142.07(s),140.08(s),134.16(s),131.62–131.13(m),130.99(s),130.71(s),127.77(d,j=4.3hz),126.69(s),120.51(s),93.93(s),83.74(s),82.26(s),77.28(d,j=11.5hz),77.02(s),76.70(s),65.60(s),31.48(s),29.69(s),24.96(d,j=13.7hz).maldi-tofmass:calcd.forc37h37bo3[m]+:m/z=540.2836;found:540.1307.
实施例10
化合物(3)的合成
在25ml的斜两口瓶中依次加入化合物4(100mg,0.12mmol,1eq.)和化合物5(150mg,0.276mmol,2.3eq.),氩气保护下加入pd(pph3)4(30mg,0.024mmol,20%),氩气氛围下继续加入冷冻除氧的1,4-二氧六环20ml,冷冻除氧的2mol/lna2co3水溶液(0.66ml,10eq),回流48h后停止。待反应液冷却至室温后,旋蒸除去溶剂,dcm萃取三次,无水硫酸镁干燥,过滤,浓缩,粗产物经硅胶层析柱提纯(洗脱剂:ea/pe=1/4),得浅黄色固体,经gpc再次提纯得82mg,产率45%。1hnmr(400mhz,cdcl3)δ9.21(s,1h),9.02(s,4h),8.20(s,2h),7.41(d,j=8.3hz,4h),7.23–7.10(m,20h),7.10–6.99(m,12h),4.27(d,j=5.5hz,2h),3.65–3.56(m,4h),3.44(t,j=6.6hz,4h),2.46(dt,j=11.5,5.7hz,1h),1.60(s,12h),1.48–1.04(m,40h),0.86(t,j=6.9hz,6h).13cnmr(101mhz,cdcl3)δ131.57–131.08(m),127.91(d,j=7.8hz),77.29(d,j=11.5hz),77.03(s),76.71(s),31.49(s),29.56(dd,j=22.4,10.3hz),22.70(s).maldi-tofmass:calcd.forc104h114n4o5[m]+:m/z=1498.8784;found:1498.1211.
实施例11
化合物(1)的合成与分离
在25ml的斜两口瓶中依次加入化合物3(117mg,0.078mmol,1eq)和naoh(624mg,15.6mmol,200eq),氩气保护下加入甲苯30ml,升高温度至回流,tlc监控反应,12h后停止反应,冷却至室温,旋蒸除去甲苯,dcm萃取三次,无水硫酸镁干燥,过滤,浓缩,粗产物经硅胶层析柱提纯(洗脱剂:ea/pe=1/10),得浅黄色固体,经质谱确认并直接用于下一步反应。在250ml的斜两口瓶中依次加入化合物2(88mg,0.064mmol,1eq),cucl(353mg,3.56mmol,56eq),cucl2(86.4mg,0.64mmol,10eq),氩气保护下加入无水吡啶80ml,60℃反应5d,冷却至室温,旋蒸除去吡啶,dcm萃取三次,无水硫酸镁干燥,过滤,浓缩,粗产物经硅胶层析柱提纯(洗脱剂:ea),经gpc再次提纯,得黄绿色固体46mg,产率52%。1hnmr(400mhz,cd2cl2)δ9.07(s,2h),8.93(s,8h),8.09(d,j=1.3hz,4h),7.38(d,j=8.3hz,8h),7.20(d,j=8.4hz,8h),7.12–7.05(m,32h),7.03–6.94(m,24h),4.15(d,j=5.7hz,4h),3.51(d,j=6.0hz,8h),3.35(t,j=6.6hz,8h),2.32(dt,j=11.8,5.9hz,2h),1.25–1.11(m,80h),0.77(t,j=6.9hz,12h).
13cnmr(101mhz,cd2cl2)δ163.12(s),160.52(s),155.34(s),145.34(s),144.28(s),143.59(s),143.24(s),139.53(d,j=15.8hz),132.98(s),132.70(s),132.30(s),131.91(s),131.62(d,j=5.3hz),128.30(s),127.35(s),126.39(s),119.97(s),116.61(s),71.71(s),69.16(s),54.24(d,j=27.2hz),53.83(s),53.56(s),53.29(s),32.33(s),30.23–29.66(m),26.62(s),23.09(s),14.29(s).maldi-tofmass:calcd.forc196h200n8o6[m+h]+:m/z=2762.5662;found:2762.9666.
图3为实施例11中的产物的聚集诱导发光现象,在不同体积比例h2o/thf的荧光光谱图(a),其中h2o为不良溶剂,thf为良溶剂,随着h2o的比例的增加,溶液的荧光也逐渐增强,说明了本发明所制备的嘧啶基团刚性大环化合物具有aie效应;便携式手提紫外荧光灯(365nm)下不同比例h2o/thf的荧光(b),在紫外荧光灯下,肉眼可见随着水比例的增大,荧光增强,说明了本发明所制备的嘧啶基团刚性大环化合物具有aie效应。
图4(a)为实施例11中的产物对芳香族化合物甲苯有一定的响应性,甲苯可以使实施例11中的产物的荧光光谱强度发生变化,并发生蓝移。图4(b)、(c)、(d)为图4(a)的光谱变化分解图。其中图4(b)中,随着甲苯量的增加,荧光强度出现下降,并发生蓝移。图4(c)中,随着甲苯量的增加,荧光强度出现增强,并继续发生蓝移。图4(d)中,随着甲苯量的继续增加,荧光强度出现减弱,并发生微弱的蓝移。
- 还没有人留言评论。精彩留言会获得点赞!