化合物lithocarolsA-F及其制备方法和在制备抗肿瘤药物中的应用与流程
本发明属于医药生物技术领域,具体涉及化合物lithocarolsa-f及其制备方法和在制备抗肿瘤药物中的应用。
背景技术:
癌症正严重威胁着人类的健康。目前,全球每年死于癌症的人数约为1000万,我国每年死于癌症的人数为250万人左右,而且这个数字还在逐年增长。当前,抗肿瘤药物的市场份额也在不断扩大,抗癌药物占医药市场份额高达30%。然而,目前抗癌药物大多存在特异性差、毒副作用强和价格昂贵等缺陷,使得癌症患者常因药物昂贵而放弃治疗或副作用大而意外致死。因此,提高抗癌药物特异性从而降低毒副作用将造福癌症患者,对人类健康和社会发展有着十分重要的现实意义。
深海来源的真菌比其他海洋微生物具有代谢产物丰富、产量大、结构类型多样及活性显著等优点,已成为海洋药物研发的热点。过去的几十年,大量的新天然产物从海洋微生物中被发现,其中部分化合物作为先导化合物已经吸引了大量有机合成化学家和药物化学家的关注,部分经半合成衍生生化的化合物已经进入临床药物实验。因此,从海洋微生物中发现新的独特结构的天然产物具有重要意义和经济价值。
技术实现要素:
本发明的第一个目的是提供具有抗肿瘤活性的化合物lithocarolsa-f。
本发明的化合物lithocarolsa-f,其结构如式(i)所示:
其中,1为化合物lithocarola、2为化合物lithocarolb、3为化合物lithocarolc、4为化合物lithocarold、5为化合物lithocarole、6为化合物lithocarolf。
本发明的第二个目的是提供一种化合物lithocarolsa-f的制备方法,该制备方法中所述的化合物lithocarolsa-f是从海洋真菌phomopsislithocarpusfs508的发酵培养物中分离制备得到的,具体包括以下步骤:
(1)制备海洋真菌phomopsislithocarpusfs508的固体发酵培养物,用乙酸乙酯萃取该固体发酵培养物,将乙酸乙酯萃取液经浓缩后得浸膏,将浸膏用甲醇水溶液分散,再用石油醚萃取,萃取剩下的甲醇水溶液部分蒸馏浓缩得到粗提物;
(2)将粗提物过硅胶柱层析,用石油醚-乙酸乙酯以体积比为10:1,7:1,9:2,2:1,1:1,0:1和二氯甲烷-甲醇以体积比为5:1,0:1分别作为洗脱剂,梯度洗脱,收集石油醚-乙酸乙酯体积比2:1洗脱获得的且经tlc薄层层析以正己烷:乙酸乙酯=5:1v/v展开得rf=0.2-0.4的组分fr.6,收集石油醚-乙酸乙酯体积比1:1洗脱获得的且经tlc薄层层析以正己烷:乙酸乙酯=2:1v/v展开得rf=0.2-0.5的组分fr.7;
将组分fr.6再经凝胶柱层析sephadexlh-20,以二氯甲烷-甲醇体积比1:1作为洗脱剂洗脱,收集tlc薄层层析以正己烷:乙酸乙酯=2:1v/v展开得rf=0.5-0.8的组分fr.6.2,将组分fr.6.2再经硅胶柱层析,以石油醚-乙酸乙酯的体积比为6:1,3:1,2:1,1:1,1:2,0:1进行梯度洗脱,收集石油醚-乙酸乙酯体积比6:1洗脱的组分fr.6.2.1,将组分fr.6.2.1用hplc分离纯化,分别得到化合物lithocarolsa-e;
将组分fr.7再经c18反相柱层析,以甲醇-水的体积比为50:50,60:40,70:30,80:20,90:10,100:0进行梯度洗脱,收集甲醇-水体积比90:10洗脱组分,得到组分fr.7.6,fr.7.6再经硅胶柱层析,以石油醚-乙酸乙酯的体积比为6:1,3:1,2:1,1:1,1:2,0:1进行梯度洗脱,收集石油醚-乙酸乙酯体积比6:1洗脱的组分fr.7.6.1;将组分fr.7.6.1用hplc分离纯化,得到化合物lithocarolf。
所述的将组分fr.6.2.1用hplc分离纯化具体为:将组分fr.6.2.1经全制备hplc在ymc-ods柱,流动相为体积比85:15的甲醇/水,流速为8ml/min的色谱条件下进行分离,于保留时间为16.9min收集洗脱组分,得到组分fr.6.2.1.5,将fr.6.2.1.5经进一步的半制备hplc,使用chiralpakic柱,流动相为体积比92:8的正己烷/异丙醇,流速为3ml/min,收集保留时间为8.1min的洗脱组分,得到化合物lithocarola,收集保留时间为9.4min的洗脱组分,得到化合物lithocarolb,收集保留时间为14.2min的洗脱组分,得到组分fr.6.2.1.3,将fr.6.2.1.3经进一步的半制备hplc,使用ymcpackods-a/aq柱,流动相为体积比85:15的乙腈/水,流速为3ml/min,收集保留时间为10.6min的洗脱组分,得到化合物lithocarolc,收集保留时间为14.8min的洗脱组分,得到组分fr.6.2.1.4,将fr.6.2.1.4经进一步的半制备hplc,使用ymcpackods-a/aq柱,流动相为体积比85:15的甲醇/水,流速为3ml/min,收集保留时间为11.7min的洗脱组分,得到化合物lithocarold,收集保留时间为18.0min的洗脱组分,得到化合物lithocarole;
所述的将组分fr.7.6.1用hplc分离纯化具体为:将组分fr.7.6.1经全制备hplc在ymc-ods柱,流动相为体积比85:15的甲醇/水,流速为8ml/min的色谱条件下进行分离,收集保留时间为16.9min的洗脱组分,得到组分fr.7.6.1.1;将fr.7.6.1.1经进一步的半制备hplc,使用chiralpakic柱,流动相为体积比95:5的正己烷/异丙醇,流速为3ml/min,收集保留时间为14.9min的洗脱组分,得到化合物lithocarolf。
所述的制备海洋真菌phomopsislithocarpusfs508的固体发酵培养物具体步骤为:挑取fs508的菌丝接种于马铃薯葡萄糖液体培养基中,在28℃、120r/min条件下培养5天,制得种子液,然后将种子液按0.1ml/g的接种量接种于大米培养基中,28℃条件下培养30天,制得fs508的固体发酵培养物,所述的马铃薯葡萄糖液体培养基,每升是通过以下方法配制的:用500ml的纯水煮200g的马铃薯,煮沸20min,过滤得马铃薯汁,再加入葡萄糖20g、kh2po43g、mgso41.5g、维生素b110mg,用水补足至1000ml,灭菌制得;所述的大米培养基是通过以下方法配制的:按每480g大米与600ml质量体积比为0.5%g/ml的粗海盐水溶液混合灭菌制得。
本发明的第三个目的是提供化合物lithocarolsa、b、c、d、e和/或f,或其药用盐在制备抗肿瘤药物中的应用。所述的抗肿瘤药物优选为抗肝癌、乳腺癌、神经胶质瘤或非小细胞肺癌药物。
本发明通过实验发现,化合物lithocarolsa-f对肝癌细胞hepg-2、乳腺癌细胞mcf-7、神经胶质瘤细胞sf-268和非小细胞肺癌细胞a549的ic50值范围为10.5~87.7μm,见表1。阳性对照物顺铂对以上四种肿瘤细胞株的ic50值分别为2.4、3.2、3.3和1.6μm。此结果表明:本发明的化合物lithocarolsa-f均具有比较显著的抗肿瘤活性。
表1化合物lithocarolsa-f的对癌细胞的抑制效果
本发明的第四个目的是提供一种抗肿瘤药物,该抗肿瘤药物包含化合物lithocarolsa-f中的至少一种,或其药用盐作为活性成分,优选,所述的抗肿瘤药物为抗肝癌、乳腺癌、神经胶质瘤或非小细胞肺癌药物。
本发明的第五个目的是提供海洋真菌phomopsislithocarpusfs508在制备化合物lithocarolsa、b、c、d、e和/或f中的应用。
与现有技术相比,本发明的优势在于:
本发明从海洋真菌phomopsislithocarpusfs508中分离制备得到化合物lithocarolsa-f,该类化合物lithocarolsa-f具有比较显著的抗肿瘤活性,可以用于制备抗肿瘤药物,为研究与开发新的抗肿瘤药物提供了候选化合物,为开发利用海洋微生物来源的天然活性物质提供了科学依据。
本发明的海洋真菌phomopsislithocarpusfs508于2018年8月15日保藏于广东省微生物菌种保藏中心(gdmcc),地址:广州市先烈中路100号大院59号楼5楼,其保藏编号为gdmccno.60433。
附图说明
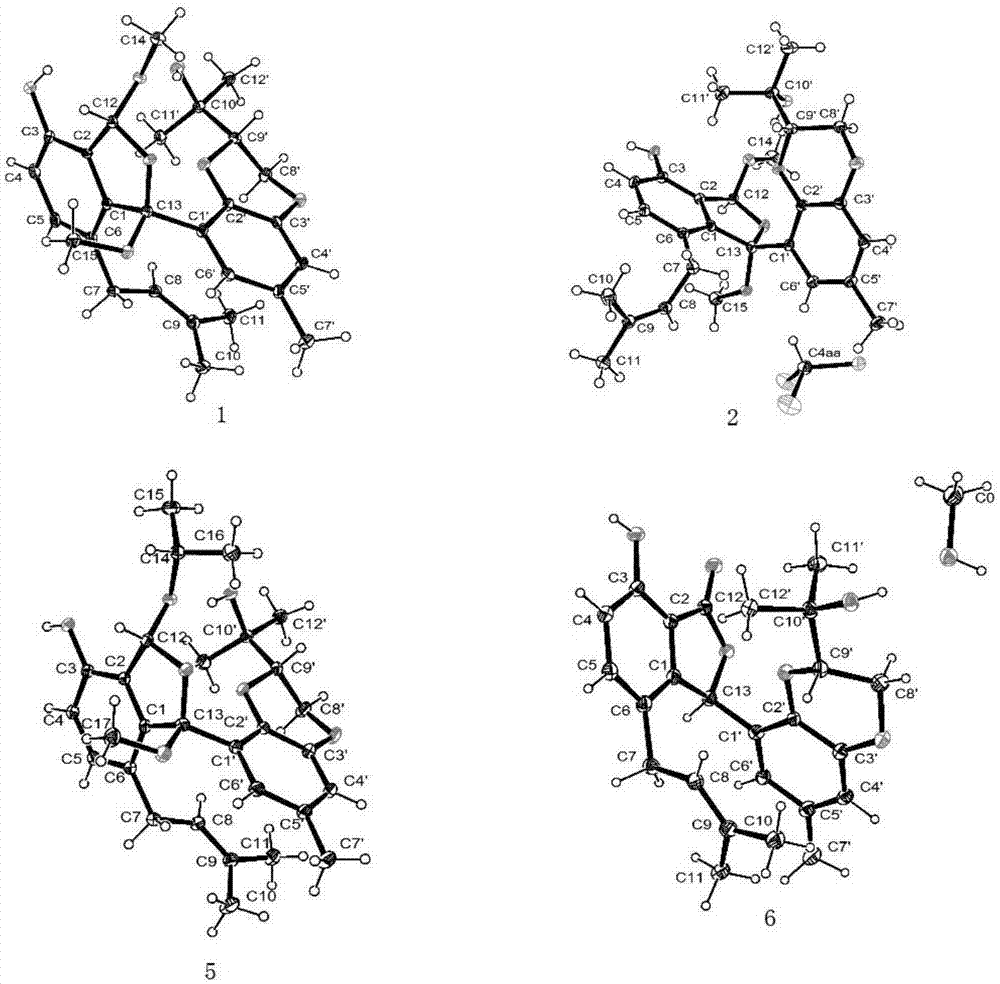
图1是化合物lithocarolsa-b和e-f的晶体结构;
图2是化合物1(lithocarola)的cosy谱;
图3是化合物1(lithocarola)的hsqc谱;
图4是化合物1(lithocarola)的hmbc谱;
图5是化合物1(lithocarola)的hr-esims谱;
图6是化合物2(lithocarolb)的cosy谱;
图7是化合物2(lithocarolb)的hsqc谱;
图8是化合物2(lithocarolb)的hmbc谱;
图9是化合物2(lithocarolb)的hr-esims谱;
图10是化合物3(lithocarolc)的cosy谱;
图11是化合物3(lithocarolc)的hsqc谱;
图12是化合物3(lithocarolc)的hmbc谱;
图13是化合物3(lithocarolc)的hr-esims谱;
图14是化合物4(lithocarold)的cosy谱;
图15是化合物4(lithocarold)的hsqc谱;
图16是化合物4(lithocarold)的hmbc谱;
图17是化合物4(lithocarold)的hr-esims谱;
图18是化合物5(lithocarole)的cosy谱;
图19是化合物5(lithocarole)的hsqc谱;
图20是化合物5(lithocarole)的hmbc谱;
图21是化合物5(lithocarole)的hr-esims谱;
图22是化合物6(lithocarolf)的cosy谱;
图23是化合物6(lithocarolf)的hsqc谱;
图24是化合物6(lithocarolf)的hmbc谱;
图25是化合物6(lithocarolf)的hr-esims谱;
具体实施方式
以下实施例是对本发明的进一步说明,而不是对本发明的限制。
实施例1
1、海洋真菌phomopsislithocarpusfs508的分离纯化和鉴定
本发明的海洋真菌phomopsislithocarpusfs508是2016年5月,从印度洋深海沉积物(16°50.508'n,111°53.335'e,水深3606米)中分离得到的,经its序列分析鉴定,genbank基因登录号为:mg686131,经blast比对,同源分析,鉴定该菌株属于phomopsis属真菌,命名为phomopsislithocarpusfs508,其于2018年8月15日保藏于广东省微生物菌种保藏中心(gdmcc),地址:广州市先烈中路100号大院59号楼5楼,其保藏编号为gdmccno.60433。
2、phomopsislithocarpusfs508的固体发酵
将活化的深海真菌fs508菌丝接种于马铃薯葡萄糖液体培养基(每升培养基是通过以下方法配制的:用500ml的纯水煮200g的马铃薯,煮沸20min,过滤得马铃薯汁,再加入葡萄糖20g、kh2po43g、mgso41.5g、维生素b110mg,用水补足至1000ml,灭菌制得)中,在28℃、120r/min条件下培养5天,制得种子液,然后将种子液按0.1ml/g大米培养基的接种量接种于大米培养基(该培养基是通过以下方法配制的:将480g大米与600ml质量体积比为0.5%mg/ml的粗海盐水溶液混合,121℃高压灭菌20min,冷却,制得)中,28℃条件下培养30天,制得fs508的固体发酵培养物。
3、化合物lithocarolsa-f的制备
(1)往fs508的固体发酵培养物中加入乙酸乙酯,浸泡提取24小时,重复提取3次,提取液经浓缩后得浸膏(99.8g);浸膏经体积分数为80%的甲醇水溶液分散,再用石油醚萃取四次,除去脂溶性成分,将萃取剩下的甲醇水溶液部分蒸馏浓缩得到粗提物。
(2)将步骤(1)得到的粗提物过硅胶柱层析,用石油醚-乙酸乙酯以体积比为10:1,7:1,9:2,2:1,1:1,0:1和二氯甲烷-甲醇以体积比为5:1,0:1分别作为洗脱剂,梯度洗脱,收集流分经tlc板合并相同主点得到14个组分,分别为fr.1-fr.14;
将组分fr.6(fr.6为石油醚:乙酸乙酯=2:1v/v洗脱获得的流分经进一步tlc薄层层析以正己烷:乙酸乙酯=5:1v/v展开得rf=0.2-0.4的组分)再经凝胶柱层析sephadexlh-20,以二氯甲烷-甲醇体积比1:1作为洗脱剂洗脱,经tlc板合并主点得到3个亚组分,分别为fr.6.1-fr.6.3;其中,得到的亚组分fr.6.2(fr.6.2为tlc薄层层析以正己烷:乙酸乙酯=2:1v/v展开得rf=0.5-0.8的组分)的重量为2.2g,将亚组分fr.6.2再经硅胶柱(200-300目)层析,以石油醚-乙酸乙酯的体积比为6:1,3:1,2:1,1:1,1:2,0:1梯度洗脱得到5个组分,分别为fr.6.2.1-fr.6.2.5;其中,得到的组分fr.6.2.1(fr.6.2.1为石油醚:乙酸乙酯=6:1v/v洗脱得到的组分)的重量为1.2g,将组分fr.6.2.1经全制备hplc在ymc-ods柱,流动相为体积比85:15的甲醇/水,流速为8ml/min的色谱条件下得到四个组分,分别为fr.6.2.1.1-fr.6.2.1.4;组分fr.6.2.1.5(保留时间为16.9min)经进一步的半制备hplc,在chiralpakic柱,流动相为体积比92:8的正己烷/异丙醇,流速为3ml/min的色谱条件下,收集保留时间为8.1min的组分,得到化合物1(化合物lithocarola,19.8mg),收集保留时间为9.4min的组分,得到化合物2(化合物lithocarolb,20.6mg),收集保留时间为14.2min的组分,得到组分fr.6.2.1.3;将fr.6.2.1.3经进一步的半制备hplc,在ymc-packods/aq柱,流动相为体积比85:15的乙腈/水,流速为3ml/min的色谱条件下,收集保留时间为10.6min的组分,得到化合物3(化合物lithocarolc,8.6mg),收集保留时间为14.8min的组分,得到组分fr.6.2.1.4,将fr.6.2.1.4经进一步的半制备hplc,在ymc-packods/aq柱,流动相为体积比85:15的甲醇/水,流速为3ml/min的色谱条件下,收集保留时间为11.7min的组分,得到化合物4(化合物lithocarold,8.6mg),收集保留时间为18.0min的组分,得到化合物5(化合物lithocarole,2.0mg)。
收集石油醚-乙酸乙酯体积比1:1洗脱获得的且经tlc薄层层析以正己烷:乙酸乙酯=2:1v/v展开得rf=0.2-0.5的组分fr.7,将组分fr.7(12.6g)再经c18反相柱层析,以甲醇-水的体积比为50:50,60:40,70:30,80:20,90:10,100:0进行梯度洗脱,收集甲醇-水体积比90:10洗脱组分,得到组分fr.7.6(2.5g),将fr.7.6再经硅胶柱层析,以石油醚-乙酸乙酯的体积比为6:1,3:1,2:1,1:1,1:2,0:1进行梯度洗脱,收集石油醚-乙酸乙酯体积比6:1洗脱的组分fr.7.6.1。将组分fr.7.6.1(0.3g)经全制备hplc在ymc-ods柱,流动相为体积比85:15的甲醇/水,流速为8ml/min的色谱条件下进行分离,得到两个主要组分fr.7.6.1.1(保留时间为16.9min)和fr.7.6.1.2(保留时间为19.8min)。将fr.7.6.1.1经进一步的半制备hplc,使用chiralpakic柱,流动相为体积比95:5的正己烷/异丙醇,流速为3ml/min,收集保留时间为14.9min的组分,得到化合物6(化合物lithocarolf,3.0mg)。
4、化合物lithocarolsa-f的结构鉴定
1h-nmr、13c-nmr、hmbc核磁共振谱图用brukeradvance-500和brukeradvance-600核磁共振光谱仪测定,以四甲基硅烷(tms)为内标;esi-ms数据用vgautospec-3000型质谱仪测定;紫外光谱用岛津uv-2600分光光度计测定,其结构鉴定如下:
如图1-25所示,图1是化合物lithocarolsa-b和d-f的晶体结构。
图2是化合物1的cosy谱;图3是化合物1的hsqc谱;图4是化合物1的hmbc谱;图5是化合物1的hr-esims谱。
图6是化合物2的cosy谱;图7是化合物2的hsqc谱;图8是化合物2的hmbc谱;图9是化合物2的hr-esims谱。
图10是化合物3的cosy谱;图11是化合物3的hsqc谱;图12是化合物3的hmbc谱;图13是化合物3的hr-esims谱。
图14是化合物4的cosy谱;图15是化合物4的hsqc谱;图16是化合物4的hmbc谱;图17是化合物4的hr-esims谱;。
图18是化合物5的cosy谱;图19是化合物5的hsqc谱;图20是化合物5的hmbc谱;图21是化合物5的hr-esims谱。
图22是化合物6的cosy谱;图23是化合物6的hsqc谱;图24是化合物6的hmbc谱;图25是化合物6的hr-esims谱。
化合物1为白色针状晶体;根据hresims[m+na]+m/z493.2197,c27h34nao7,计算值为493.2197,确定化合物的分子式为c27h34o7,不饱和度为9;13c-nmr谱显示有27个碳信号(表2),包括7个甲基,2个亚甲基,7个次甲基以及11个季碳,进一步通过分析化合物1的二维谱图和x-ray晶体衍射实验确定了化合物1的结构。
化合物2为白色针状晶体(其核磁数据如表2所示);根据高分辨质谱(hresims)[m+na]+m/z493.2203,c27h34nao7,计算值为493.2197,确定化合物的分子式为c27h34o7,不饱和度为9;通过分析其1d和2d谱图发现化合物2的谱图与化合物1的谱图具有极大的相似度,化合物2应具有化合物1相同的母核结构,通过仔细分析化合物2的hmbc和cosy相关谱图推测出化合物2为化合物1的差向异构体。最终,通过x-ray晶体衍射实验确定了化合物2的结构。
表2化合物1和2的核磁数据(cd3cood3,600mhz/150mhz,δinppm,jinhz)
化合物3为黄色油状物(其核磁数据如表3所示);化合物3的分子量与化合物1相同;通过分析其1d和2d谱图发现化合物2的谱图与化合物1的谱图具有极大的相似度,化合物2应具有化合物1相同的母核结构,通过仔细分析化合物2的hmbc和cosy相关谱图推测出化合物3为化合物1的同分异构体。确定其平面结构后,进一步通过ecd计算确定了其绝对构型。
化合物4为黄色油状物(其核磁数据如表3所示);根据高分辨质谱(hresims)[m+na]+m/z493.2200,c27h34nao7,计算值为493.2197,确定化合物的分子式为c27h34o7,不饱和度为9;通过分析其1d和2d谱图发现化合物4的谱图与化合物3的谱图具有极大的相似度,化合物4应具有化合物3相同的母核结构,通过仔细分析化合物4的hmbc和cosy相关谱图推测出化合物4为化合物3的差向异构体。确定其平面结构后,进一步通过ecd计算确定了其绝对构型。
表3化合物3和4的核磁数据(cd3cood3,1h-nmr(500mhz)和13c-nmr(125mhz))
化合物5为白色针状晶体(其核磁数据如表4所示);根据高分辨质谱(hresims)[m-h]–m/z497.2550,c29h37o7,计算值为497.2545,确定化合物的分子式为c29h38o7,不饱和度为11;通过分析其1d和2d谱图发现化合物5的谱图与化合物1的谱图具有极大的相似度,主要的区别是化合物1中5位的甲氧基在化合物5中为一个异丙基所取代,通过仔细分析化合物5的hmbc和cosy相关谱图和x-ray晶体衍射实验,最终确定了化合物5的结构。
化合物6为白色针状晶体(其核磁数据如表4所示);根据高分辨质谱(hresims)[m+cl]–m/z459.1582,c25h28clo6,计算值为459.1580,确定化合物的分子式为c29h38o7,不饱和度为10;通过分析其1d和2d谱图发现化合物6的谱图与化合物1的谱图具有极大的相似度,主要的区别是化合物6形成了一个新的内酯环,而存在于化合物1中的两个甲氧基在化合物6中消失了。通过仔细分析化合物6的hmbc和cosy相关谱图和x-ray晶体衍射实验,最终确定了化合物6的结构。
表4化合物5和6的核磁数据(cd3cood3,1h-nmr(500mhz)和13c-nmr(125mhz))
经过上述方法分离的目标化合物1命名为化合物lithocarola,化合物2命名为化合物lithocarolb,化合物3命名为化合物lithocarolc,化合物4命名为化合物lithocarold,化合物5命名为化合物lithocarole,化合物6命名为化合物lithocarolf,化合物lithocarolsa-f的结构式如式(i)所示:
实施例2
采用srb法(skehanp,storengr,dominics.newcolorimetriccytotoxicityassayforanticancer-drugscreening[j].jnatlcanceinst,1990,82:1107–1112.)测试化合物lithocarolsa-f的抗肿瘤活性。
1、试验用试剂:将本发明制备的化合物lithocarolsa-f用二甲基亚砜(dmso)溶解得浓度为10mg/ml的母液,再用rpmi-1640培养基稀释至所需浓度。阳性对照为顺铂水溶液。
本实验所用肿瘤细胞株为肝癌细胞hepg-2、乳腺癌细胞mcf-7、神经胶质瘤细胞sf-268和非小细胞肺癌细胞a549。
2、实验方法:取对数生长期的hepg-2、mcf-7、sf-268和a549细胞,用胰酶消化,台盼蓝染色计数,台盼蓝排斥实验检测细胞活力大于95%后,用新鲜rpmi-1640培养基调整细胞浓度为3×104个/ml,细胞接种于96孔板,每孔加入180μl的细胞悬液,并设3个空白孔调零,于37℃、5%co2培养箱培养24h。待细胞贴壁后,每孔加入20μl一定浓度的上述化合物lithocarolsa-f的溶液,阴性对照加20μlrpmi-1640培养基,以顺铂作阳性对照。置于37℃、5%co2培养箱中培养72h后,加入50μl体积分数为50%的冷三氯醋酸水溶液固定细胞,4℃放置1h后用蒸馏水洗涤5次,空气中自然干燥。然后加入由体积分数1%冰醋酸水溶液配制的磺酰罗丹明b(sulforhodamineb,srb)4mg/ml溶液100μl/孔,室温中染色30min,去上清,用1%冰醋酸洗涤5次,空气干燥。最后加入200μl/孔,浓度为10mmol/ml的tris溶液,用酶标仪测定570nm处的吸光值(a),用以下公式计算药物对细胞生长的抑制率:细胞生长抑制率(%)=(1-a样品组/a对照组)×100%。
3、实验结果:本发明制备的化合物lithocarolsa-f对四株肿瘤细胞的细胞毒性如表1所示。此结果表明:本发明的化合物lithocarolsa-f具有比较显著的抗肿瘤活性,因此,本发明为研究与开发新的抗肿瘤药物提供了候选化合物,为开发利用深海微生物来源的天然活性物质提供了科学依据。
表1化合物lithocarolsa-f的对癌细胞的抑制效果
以上仅是本发明的优选实施方式,应当指出的是,上述优选实施方式不应视为对本发明的限制,本发明的保护范围应当以权利要求所限定的范围为准。对于本技术领域的普通技术人员来说,在不脱离本发明的精神和范围内,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!