一种改进的16S-seq方法及其应用与流程
本发明涉及植物附属微生物组分析技术领域,具体涉及一种改进的16s-seq方法及其应用。
背景技术:
植物根系微生物主要分为三部分,根际,根围以及根系内生菌。富集在根系周围的微生物大多数对植物的生长发育是有益的,它们不仅能提高植物的抗病性、从土壤中获取养分的能力,而且还能分泌有益物质促进植物的生长发育。完整和部分的16srrna基因测序的方法已经成为鉴定微生物种类的有用工具,尤其是近年来发展迅速的高通量测序技术,具有速度快、成本低、覆盖度深、产出巨大等优势。16srrna位于原核细胞核糖体小亚基上,微生物16srrna基因包含9个“高度可变区”(hypervariableregions),在不同的微生物中具有相当大的多样性,16s小亚基核糖体rna基因的高通量测序(16s-seq)技术,就是通过在16s序列保守区上设计通用引物,对一个或多个高度可变区进行扩增,然后对扩增子序列进行高通量测序,从而能够快速的鉴定出不同的微生物种类,植物附属微生物群落的结构和动态的研究就是主要运用这种扩增子深度测序的方法进行的。然而从宿主植物组织中扩增这些高度可变区的时候,由于宿主植物的植物质体和线粒体由原核生物进化形成,并且保留了微生物的16srrna的基因,因此,16s-seq中使用的通用引物也能扩增出宿主植物的rrna基因,从而减少了16s-seq的测序效率。例如,当对来源于植物样品中的16srrna基因v4高度可变区进行测序时,来源于宿主植物的质体和线粒体序列高达所有测序序列的95%。如此高的宿主序列污染限制了同时测序的样本数量和样本的测序深度,以及16s-seq技术在描述微生物群落结构和多样性的运用价值。因此,研究一种能有效去除宿主污染的方法对于研究宿主附属微生物群落具有重要的价值。
近年来,crispr/cas9系统作用机制的发现及其应用彻底改变了基因组编辑技术,cas9内切酶在grna的引导下能够快速、精确且高效地切断双链dna分子,形成双链dna断裂(double-strandeddnabreak,dsb)。crispr/cas9系统主要是由规律成簇的间隔短回文重复序列(crispr)与cas9蛋白组成。利用cas9/grna标靶特定的dna位点只需满足2个条件:(1)grna的5’端20nt(nucleotides)的引导序列(称为spacer或guidesequence)与靶dna位点的序列(称为protospacer)互补匹配;(2)靶位点的必需存在pam(protospacer-adjacentmotif),其中使用最广的化脓链球菌cas9的pam序列为5’-ngg-3’。cas基因编码的蛋白能够在sgrna的引导下特异性切割靶位点。但是crispr/cas9与靶位点识别的特异性主要依赖于grna与靠近pam处10-12bp碱基的配对,而其余远离pam处8-10bp碱基的错配对靶位点的识别影响不明显,说明了crispr/cas9存在严重的脱靶性,本发明主要就是利用crispr/cas9系统的这些特点,结合crispr/cas9系统的脱靶原则设计出特异性的grnaspacer序列,合成grna序列,使其能够引导cas9蛋白体外特异性的剪切的寄主植物rrna序列,而不剪切的微生物rrna序列。
技术实现要素:
本发明的目的在于提供一种特异性grnaspacer序列及利用crispr/cas9系统特异性剪切构建植物附属微生物扩增子文库中产生的宿主植物rrna序列的方法。本发明提供的特异性grnaspacer序列形成的grna序列能够引导cas9实现水稻rrna序列的特异性切割,实现微生物rrna的富集,这种改进的16s-seq方法为分析植物附属微生物组的研究奠定了基础。
本发明提供了一种利用crispr/cas9系统特异性剪切水稻rrna序列的grnaspacer序列,所述grnaspacer的核苷酸序列如seqidno.1~16任意一条序列所示。
本发明提供了一种利用crispr/cas9系统特异性剪切水稻rrna序列的grna序列,所述grna序列由上述grnaspacer序列的任意一条与grnascaffold序列组成。
优选的,所述grnascaffold的序列如seqidno.17所示。
本发明以剪切16s-seq测序文库中的产生水稻rrna序列为例,提供了一种利用crispr/cas9系统特异性剪切16s-seq测序文库产生的宿主植物rrna序列的方法,包括以下步骤:
1)提取水稻根系微生物dna,得到扩增模板;
2)将步骤1)得到的扩增模板与序列如seqidno.18~19所示的引物混合,采用降落pcr方法、利用16srdnabacterialindentificationpcrkit试剂盒进行第一扩增,得到第一扩增产物;
3)利用crispr/cas9系统对步骤2)得到的第一扩增产物进行剪切反应,得到剪切产物;每20μl所述剪切反应的体系包括:第一扩增产物2μl、无核酸酶水13μl、10×cas9核酸酶反应缓冲液2μl、1μm的cas9核酸酶2μl、90℃,5min变性处理的权利要求2所述grna序列的任意一条1μl(60ng);
4)将步骤3)得到的剪切产物与引物p5+bc1和p7+bc2、i5-2xhighfidelitymastermix混合,进行第二扩增,得到去除大量水稻rrna序列,大量富集的微生物rrna序列;所述引物p5+bc1为在如seqidno.20所示核苷酸序列的3’端依次修饰有拆分样品的p5端index序列和illumina高通量测序平台p5端通用的的接头序列;所述引物p7+bc2为在如seqidno.21所示核苷酸序列的3’端依次修饰有拆分样品的p7端index序列和illumina高通量测序平台p7端通用的的接头序列。
优选的,步骤2)得到第一扩增产物后,还包括对第一扩增产物进行纯化的过程,所述纯化为磁珠法纯化。
优选的,步骤3)所述剪切反应的温度为37℃,时间为16h。
优选的,步骤2)所述第一扩增的反应条件为:94℃5min;4个循环:94℃1min,60℃1min,72℃45s;6个循环:94℃1min,58℃1min,72℃45s;8个循环:94℃1min,56℃1min,72℃45s;8个循环:94℃1min,54℃1min,72℃45s;8个循环:94℃1min,52℃1min,72℃45s;72℃5min。
优选的,步骤4)所述第二扩增的反应条件为:98℃2min;14个循环:98℃30s,58℃30s,72℃15s;72℃5min。
本发明还提供了上述grnaspacer序列或grna序列或利用上述方法在利用改进的16s-seq技术分析植物附属微生物组的方法中的应用。
本发明基于crispr/cas9系统,在利用16s小亚基核糖体rna基因的高通量测序(16s-seq)技术,分析植物附属微生物群落的结构和动态测序文库制备过程中,利用设计出的特异性grna(guiderna)引导cas9切割植物宿主rrna基因,开发了一种高效的准确的16s-seq中去除宿主rrna的方法,大大降低了宿主rrna污染对16s-seq技术分析植物附属微生物群落的有效性以及其测序深度的影响,并且这种方法能广泛的用于植物附属微生物组分析中。
在本发明实施例中,为了检验这种改进的16s-seq的方法在去除宿主rrna的效率,首先用不同比例(0.1%-50%)混合的人工合成的水稻和微生物rrna样品,检验了cas9介导的去除宿主rrna的效率。发现这种新方法可以有效地从混合样品中去除几乎所有的水稻rrna。然后为了验证cas9/grna的处理不会对16s-seq测序结果产生偏好性影响,使用16s-seq(cas9处理/不处理)对田间土壤微生物群体进行了分析,结果表明,cas9处理不会引入对微生物群体分析的任何偏好性。最后,利用改进的16s-seq的方法分析了水稻根系样品。cas9的处理能去除80%以上的水稻rrna,极大的增加了测序观察到的微生物种类的数量。综上所述,本发明开发了一种高效的准确的16s-seq中去除宿主植物rrna的方法,并且这种方法能广泛的用于植物微生物组分析中。
附图说明
图1为本发明提供的改进的16s-seq的方法构建植物附属微生物群体测序文库的流程;
图2为本发明结合crispr/cas9系统的脱靶原则挑选出特异性的grnaspacer序列;
图3为本发明设计带有黏性末端以及grnaspacer的引物原理图;
图4为本发明实施例1中评估合成的高特异性grna引导cas9体外剪切宿主植物rrna基因扩增子的活性;
图5为本发明检测改进的16s-seq的方法富集微生物rrna的效率,并与传统pnapcr钳的方法进行比较;
图6为本发明利用改进的16s-seq技术构建的水稻土壤微生物样品16s-seq测序文库;
图7为本发明实施例3中基于otus的cas9/grna处理组和未经cas9/grna处理对照组venn图结果;
图8为本发明实施例3中基于otu出现频率的相关性分析;
图9为本发明cas9/grna处理组和未经cas9/grna处理对照组的测序结果upgma聚类树分析;
图10为本发明利用改进的16s-seq构建水稻根系样品16s-seq测序文库;
图11为本发明实施例4中基于otus的cas9/grna处理组和未经cas9/grna处理对照组venn图结果;
图12为本发明于实施例4中不同处理组共有otu出现频率的相关性分析;
图13为本发明cas9/grna处理组和未经cas9/grna处理对照组测序样品的稀释曲线分析;
图14为本发明cas9/grna处理组和未经cas9/grna处理对照组的测序结果upgma聚类树分析。
具体实施方式
本发明提供了一种利用crispr/cas9系统特异性剪切水稻rrna序列的grnaspacer序列,所述grnaspacer的核苷酸序列如seqidno.1~16任意一条序列所示。本发明所述grnaspacer序列是利用已发表微生物16srrna高度可变区(v1-v2区、v4区和v5-v7区)所有已知序列与共扩增的寄主植物(水稻)器官rrna序列比对挑选出的。本发明所述比对挑选方法如图2所示:利用rdp数据库(https://rdp.cme.msu.edu/)中已发表微生物16srrna所有已知序列与寄主植物质体和线粒体细胞器rrna序列,找出所有潜在的grnaspacer序列,然后进行全局成对比对,结合crispr/cas9系统的脱靶原则挑选特异性的grnaspacer序列,使利用其合成的grna引导cas9只剪切宿主植物的rrna序列,而不剪切微生物的rrna序列。本发明所述grnaspacer序列合成的grna能引导cas9只剪切寄主植物(水稻)的rrna序列,而不剪切微生物rrna序列,具有高特异性,具体序列如表1所示:在v1-v2区共挑选出4个特异性的grnaspacer序列,v4区共挑选出5个特异性的grnaspacer序列,在v5-v7区共计挑选出7个特异性的grnaspacer序列。
表1不同高度可变区扩增子区域筛选出的grnaspacer序列
本发明还提供了一种利用crispr/cas9系统特异性剪切水稻rrna序列的grna序列,所述grna序列由上述技术方案所述grnaspacer序列的任意一条与grnascaffold序列组成。在本发明中,所述grnascaffold的序列如seqidno.17所示:gttttagagctagaaatagcaagttaaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgc。本发明对所述grna序列的合成方法没有特殊的限定,具体地,本发明体外合成带有特异性grnaspacer的grna序列的方法包括以下步骤:
①将puc19质粒载体的bsai酶切位点进行突变将形成的载体命名为(puc19-bsai突);
②从载体prgeb32扩增出带有t7启动子、bsai双酶切位点的grnascaffold序列;所述扩增用引物如seqidno.23~25所示;
③将步骤②得到的带有t7启动子、bsai双酶切位点的grnascaffold序列采用乙醇醋酸钠回收,并利用kpni+bamhi进行双酶切;
④将步骤①得到的puc19-bsai突载体利用kpni+bamhi进行双酶切,胶回收得到酶切后的puc19-bsai突载体片段;
⑤利用t4dna连接酶将带有t7启动子、bsai双酶切位点的grnascaffold序列与酶切后的puc19-bsai突载体片段进行连接,利用dh5α大肠杆菌菌株进行转化,进行菌落pcr验证;
⑥利用引物m13f和m13r进行测序(见表3),测序结果正确,载体质粒命名为puc19-sgrna;
⑦设计带有黏性末端以及grnaspacer的引物序列;
⑧将引物进行磷酸化并利用引物退火形成带有bsai双酶切位点以及特异性grnaspacer的片段;
⑨将带有bsai双酶切位点以及特异性grnaspacer的片段利用bsai进行酶切,将步骤⑥得到的puc19-sgrna载体利用bsai进行酶切,并用去磷酸化酶进行处理,利用t4dna连接酶将带有bsai双酶切位点以及特异性grnaspacer的片段与酶切后的puc19-sgrna载体进行连接,利用dh5α大肠杆菌菌株进行转化,进行菌落pcr验证,挑选正确菌落进行培养,提取质粒;将正确质粒命名为puc19-osgrna(序列如seqidno.59所示);
⑩利用引物m13f和grna-r从puc19-osgrna上扩增出带有t7启动子、特异性的grnaspacer和grnascaffold序列的片段;
所述步骤②和①没有时间先后顺序的限定;
所述步骤⑦和步骤①~⑥没有时间先后顺序的限定。
本发明将puc19质粒载体的bsai酶切位点进行突变将形成的载体命名为(puc19-bsai突);本发明所述突变能够使载体上不再具有bsai酶切位点,有利于在第⑨步利用bsai进行酶切puc19-sgrna载体时,只剪切插入的bsai双酶切位点,而不剪切载体的其他部位。本发明对所述突变的方法没有特殊的限定,采用常规突变方法即可。本发明对所述puc19质粒载体的来源没有特殊限定,采用本领域技术人员熟知的puc19质粒载体的常规市售产品即可。
本发明从载体prgeb32扩增出带有t7启动子、bsai双酶切位点的grnascaffold序列;所述扩增用引物如seqidno.18~19所示。在本发明中,grnascaffold序列是由不包含grnaspacer的crrna以及全部tracrrna序列构成。本发明对所述载体prgeb32的来源并没有特殊限定,利用本领域的常规市售产品即可。在本发明中,所述扩增用引物名称及序列如表2所示:
表2构建特异性的grna表达载体引物序列
在本发明中,所述t7启动子序列主要是为了第
得到带有t7启动子、bsai双酶切位点的grnascaffold序列后,本发明将带有t7启动子、bsai双酶切位点的grnascaffold序列采用乙醇醋酸钠回收,并利用kpni+bamhi进行双酶切。本发明对所述乙醇醋酸钠回收的具体方法没有特殊的限定,采用本领域技术人员熟知的乙醇醋酸钠回收基因片段的常规方法即可。本发明对所述双酶切的具体条件没有特殊的限定,依据kpni和bamhi的常规双酶切条件参数即可。
本发明将得到的puc19-bsai突载体利用kpni+bamhi进行双酶切,胶回收得到酶切后的puc19-bsai突载体片段。本发明对所述双酶切和胶回收方法没有特殊的限定,采用本领域技术人员熟知的常规双酶切、胶回收方法条件即可。
利用t4dna连接酶将带有t7启动子、bsai双酶切位点的grnascaffold序列与酶切后的puc19-bsai突载体片段进行连接,利用dh5α大肠杆菌菌株进行转化,进行菌落pcr验证。本发明对所述转化方法没有特殊的限定,采用本领域技术人员熟知的质粒转化方法即可。
转化成功后,本发明利用引物m13f和m13r进行测序(见表2),测序结果正确,载体质粒命名为puc19-sgrna。在本发明中,所述m13f引物的序列为5’-ggtaacgccagggttttcc-3’(seqidno.24),所述m13r引物的序列为5’-caggaaacagctatgacc-3’(seqidno.26)。
设计产生粘性末端以及grnaspacer的引物序列,引物设计原理图如图3所示,其中图3中的tagg和caaa为了引物退火后产生黏性末端,更好与bsai酶切后载体连接,所添加的序列。n为grnaspacer序列。得到引物的具体序列如表3所示:
表3带有产生粘性末端以及grnaspacer的引物序列
本发明将引物进行磷酸化并利用引物退火形成带有黏性末端以及特异性grnaspacer的片段。在本发明中,所述退火体系包括:4.5μlf引物(100μm),4.5μlr引物(100μm)和1μlnebbuffer2。本发明所述退火优选在pcr仪上进行,设置退火程序:95℃,5min;95–85℃,-2℃/s;85–25℃,-0.1℃/s;25℃,5min。在本发明中,所述磷酸化可将片段与去磷酸化处理载体更好连接。
将步骤⑥得到的puc19-sgrna载体利用bsai进行酶切,并用去磷酸化酶进行处理,利用t4dna连接酶将带有黏性末端及特异性grnaspacer的片段与酶切后的puc19-sgrna载体进行连接,利用dh5α大肠杆菌菌株进行转化,进行菌落pcr验证,挑选正确菌落进行培养,提取质粒;将正确质粒命名为puc19-osgrna。本发明对所述转化、菌落pcr和质粒的提取方法均没有特殊限定,采用本领域技术人员熟知的常规方法即可。
得到puc19-osgrna后,本发明利用引物m13f和grna-r从puc19-osgrna上扩增出带有t7启动子、特异性的grnaspacer和grnascaffold序列的片段;在本发明中,所述引物grna-r的序列为5’-aaaagcaccgactcgg-3’(seqidno.27)。
得到带有t7启动子、特异性的grnaspacer和grnascaffold序列的片段后,本发明胶纯化带有t7启动子、特异性的grnaspacer和grnascaffold序列的片段,并利用t7quickhighyieldrnasynthesiskit(neb公司)进行体外转录,合成带有特异性grnaspacer的grna序列。
本发明还提供了上述grnaspacer序列或grna序列或利用改进的16s-seq方法在分析植物附属微生物组中的应用。
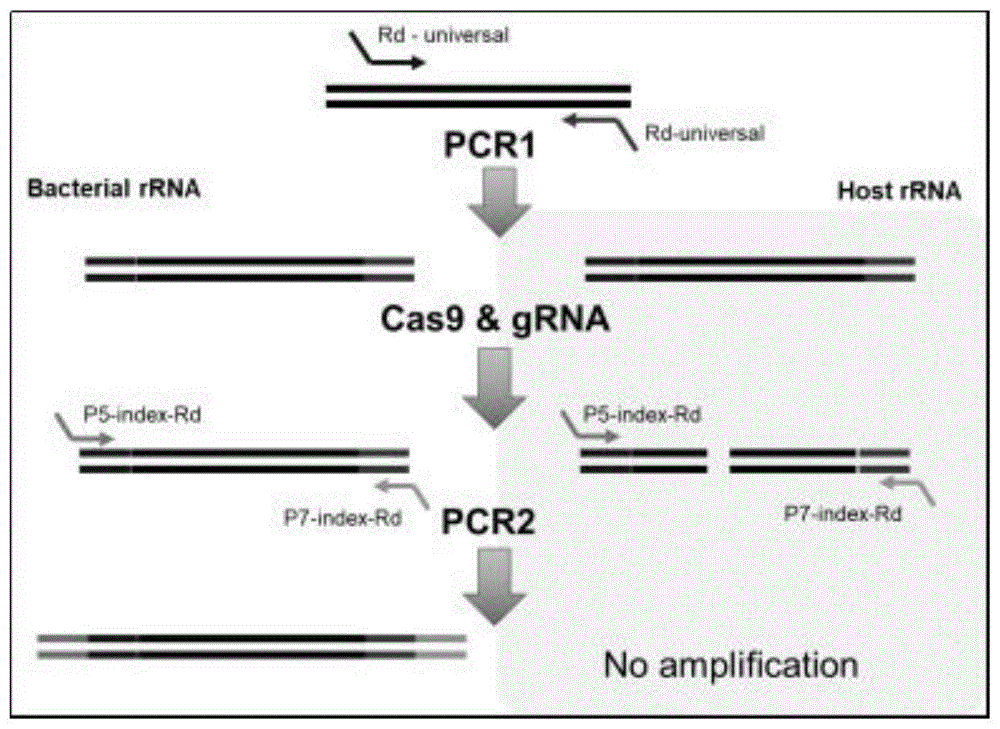
本发明利用crispr/cas9系统特异性剪切宿主植物rrna序列的方法涉及两段式扩增反应,能够得到构建扩增子文库,利用cas9/grna处理,以及两段式pcr构建扩增子文库流程图如图1所示:rd_universal/代表构建16s测序文库所用的引物,universal代表扩增16s不同高度可变区的引物序列,rd代表illumina测序平台readsequence序列。pcr1代表第一轮和第二轮扩增,扩增出目标研究区域。cas9&grna代表利用cas9/grna处理第一轮扩增产物。p5/p7-index-rd为第二轮扩增用引物序列,p5/p7代表illumina测序平台接头序列,index为拆分不同样品的标签序列,pcr2代表为第二轮扩增加上illumina测序平台接头序列。这种改进的16s-seq的方法能有效的去除构建测序文库中寄主植物rrna的序列。
本发明提取水稻根系微生物dna,得到扩增模板。在本发明中,所述水稻根系微生物dna优选利用fastdnaspinkitforsoil(mb公司)试剂盒,提取水稻根系微生物dna,所述水稻根系微生物dna中不仅含有微生物的基因组dna,还含有大量的水稻基因组dna。
得到扩增模板后,本发明将扩增模板与序列如seqidno.18~19所示的引物混合,采用降落pcr方法、利用16srdnabacterialindentificationpcrkit试剂盒进行第一扩增,得到第一扩增产物。在本发明中,所述引物如rd1+799f(seqidno.18)和rd2+1193r(seqidno.19)所示,所述引物rd1+799f和rd2+1193r为包含测序readsequencing序列和16srdnav5-v7区高度可变区扩增引物序列;引物rd1+799f的核苷酸序列为5’-tcgtcggcagcgtcagatgtgtataagagacagaacmggattagataccckg-3’,引物rd2+1193r的核苷酸序列为5’-gtctcgtgggctcggagatgtgtataagagacagcgtcatccmcaccttcctc-3’。本发明对16srdnabacterialindentificationpcrkit试剂盒的来源没有特殊的限定,优选采用takara公司生产的常规市售16srdnabacterialindentificationpcrkit试剂盒。在本发明中,所述第一轮扩增产物中含有大量的共扩增的寄主植物(水稻)rrna序列,而微生物rrna序列却很少。在本发明中,所述第一扩增的反应条件(如表4所示)为:94℃5min;4个循环:94℃1min,60℃1min,72℃45s;6个循环:94℃1min,58℃1min,72℃45s;8个循环:94℃1min,56℃1min,72℃45s;8个循环:94℃1min,54℃1min,72℃45s;8个循环:94℃1min,52℃1min,72℃45s;72℃5min;其中backto_cycles中2,14分别表示返回第2步进行14循环;5,6分别表示返回第5步进行6个循环,以此类推。
表4利用降落pcr的方法进行第一轮扩增
在本发明中,得到第一扩增产物后,还包括对第一扩增产物进行纯化的过程,所述纯化为磁珠法纯化。
得到第一扩增产物后,更优选得到纯化后的第一扩增产物后,本发明利用cas9/grna对第一扩增产物进行剪切反应,得到剪切产物;每20μl所述剪切反应的体系包括:第一扩增产物2μl、无核酸酶水13μl、10×cas9核酸酶反应缓冲液2μl、1μm的cas9核酸酶2μl、90℃,5min变性处理的权利要求2所述grna序列的任意一条1μl(60ng);所述剪切反应的时间为16h。在本发明中,步骤3)所述剪切反应的温度为37℃。本发明在进行所述剪切反应之前,优选还包括将所述无核酸酶水13μl、10×cas9核酸酶反应缓冲液2μl、1μm的cas9核酸酶2μl和grna混合后,于25℃下孵育15min。在本发明中,通过特异性grna的使用,带有特异性grnaspacer的grna能够引导cas9剪切共扩增的寄主植物(水稻)rrna序列,而不剪切第一轮扩增产物中的微生物rrna序列。在本发明中,所述剪切体系如表5所示:
表5利用crispr/cas9系统对第一扩增产物进行剪切反应体系
得到剪切产物后,本发明将剪切产物与引物p5+bc1和p7+bc2、i5-2xhighfidelitymastermix混合,进行第二扩增,得到去除大量水稻rrna序列的微生物rrna序列;所述引物p5+bc1为在如seqidno.20所示核苷酸序列的3’端依次修饰有拆分样品的p5端index序列和高通量测序的p5端接头序列;所述引物p7+bc2为在如seqidno.21所示核苷酸序列的3’端依次修饰有拆分样品的p7端index序列和高通量测序的接头序列。在本发明中,所述p5端index序列及p7端index序列如表7所示。在本发明中,所述引物p5+bc1为:aatgatacggcgaccaccgagatctacac[index1]tcgtcggcagcgtcagatgt;所述引物p7+bc2为:caagcagaagacggcatacgagat[index2]gtctcgtgggctcggagatg。在本发明中,所述index序列可在高通量测序时拆分不同样品。在本发明中,所述接头序列与第一扩增所用的引物有重合部分,在第二扩增过程中能以剪切产物为模板进行第二轮扩增。在第二扩增过程中,由于第一扩增产物中的共扩增寄主植物(水稻)rrna序列被cas9剪切,所以不能被进一步扩增,而微生物rrna序列没有被cas9剪切所以能够进行扩增,从而大量富集微生物的rrna序列。在本发明中,所述第二扩增的反应条件为:98℃2min;14个循环:98℃30s,58℃30s,72℃10min;72℃5min。
表6第二扩增的反应条件
针对不同样品加入的index序列如表7所示,其中i5表示llumina测序平台p5端加入的index序列,i7表示illumina测序平台p7端加入的index序列:
表7.不同样品加入的index序列
第一扩增和第二扩增用引物如表8所示:
表8第一扩增和第二扩增用引物(构建测序扩增子文库序列)
下面结合具体实施例对本发明所述的利用crispr/cas9系统特异性剪切水稻rrna序列的方法做进一步详细的介绍,本发明的技术方案包括但不限于以下实施例。
实施例1
体外剪切合成的宿主植物rrna基因扩增子,评估合成的高特异性grna引导cas9剪切宿主植物rrna序列的活性
1)合成引导cas9剪切叶绿体rrna扩增子和线粒体rrna扩增子的高特异性grna共计16个。
2)准备体外剪切底物。将水稻16srrna扩增子(以水稻基因组dna为模板,27f-338r、515f-806r、799f-1193r扩增),克隆到peasy-blunt载体中,然后利用m13f/m13r引物进行扩增,利用苯酚氯仿纯化扩增产物,产物即为体外剪切底物。
3)将准备好的体外剪切底物,利用cas9/grna进行体外剪切,所述剪切反应的体系如表9所示。
表9剪切反应体系
3)计算cas9/grna剪切水稻16srrna扩增子序列的效率
将步骤2)得到的水稻16srrna扩增子产物,利用步骤3)的剪切体系进行16h过夜剪切,利用2%琼脂糖凝胶进行电泳。电泳结果如图4所示,其中图4-1为利用宿主16srrna基因,在对应于微生物16s高度可变区v1-v2区筛选出的四个特异性grnaspacer序列,合成grna引导cas9体外剪切水稻叶绿体rrna扩增子序列;图4-2为利用宿主16srrna基因,在对应于微生物16s高度可变区v4区筛选出的四个特异性grnaspacer序列,合成grna引导cas9体外剪切水稻线粒体rrna基因扩增子序列。评估其引导cas9体外剪切水稻线粒体rrna基因扩增子序列的活性;图4-3为利用宿主16srrna基因,在对应于微生物16s高度可变区v5-v7区筛选出的四个特异性grnaspacer序列,合成grna引导cas9体外剪切水稻线粒体rrna基因扩增子序列;“+”代表加入cas9进行酶切处理组;“-”代表未加入cas9进行酶切处理的对照组。
通过imagej软件计算成功被剪切的条带占总rrna量(没有利用cas9/grna处理的对照组条带)的百分比,即体外剪切效率。在我们测试的16个grnas中,大部分的grna能引导cas9去除大部分的水稻rrna扩增子产物,其中有6个grnas引导cas9能100%剪切纯化的水稻rrna扩增子序列。
实施例2
利用人工合成的rrna扩增子序列样品,检测这种基于cas9/grna处理的改进的16s-seq的方法富集微生物rrna的效率,并与传统pnapcr钳的方法进行比较
1)人工合成微生物的rrna扩增子序列。利用土壤微生物dna为模板,利用799f/1193r引物进行扩增,将扩增产物克隆到peasy-blunt载体中,利用m13f/m13r引物进行扩增,利用苯酚氯仿纯化扩增产物,产物即为人工合成微生物的rrna扩增子序列。
2)将人工合成的水稻rrna扩增子序列(实施例1中第2步合成)和微生物rrna扩增子序列按照质量分数1000:1;500:1;5:1;2.5:1;1:1的比例的量添加(总量80ng),利用cas9/grna((grnaid为mito1048具体序列见交底书中表2))体外剪切,剪切体系按照表3中描述的体系配置,16h剪切处理,同时未加cas9处理为对照组,取1μl剪切产物,利用m13f/m13r引物进行扩增,2%琼脂糖凝胶电泳验证(图5)
3)利用上述2中比例混合人工合成rrna扩增子序列样品,总体积20μl。取1μl混合样品,利用pnapcr钳的方法,利用m13f/m13r引物进行pcr扩增,2%琼脂糖凝胶电泳,检测其富集微生物rrna的效率。(图5)
这种基于cas9/grna处理的改进的16s-seq的方法,能有效去除人工混合样品中所有的水稻rrna扩增子序列,并且在不同比例的混合样品中都能有效富集微生物的rrna扩增子序列。相比之下,传统的pnapcr钳的方法,只能去除部分的水稻rrna扩增子序列,并且只能在微生物rrna含量在混合样品40%-50%时,才能有效富集微生物rrna扩增子序列。
实施例3
检测基于crispr/cas9系统改进的16s-seq的方法是否会对测序结果产生偏好性影响。
利用cas9/grna处理,构建土壤微生物样品16s-seq测序文库
1.1提取土壤微生物dna(取自种植42天的mh63水稻根系周围土壤提取,不含植物组织,利用fastdnaspinkitforsoil(mb公司)试剂盒,共取三个不同植株的根系周围土壤,另外加一个空白对照)。提取水稻基因组dna(无菌培养11天的mh63植株,取三棵不同植株的叶片,外加一个空白对照)。
1.2利用改进的16s-seq的方法构建扩增子文库(附图1)。
1.3第一轮扩增(程序见表4)利用16srdnabacterialindentificationpcrkit试剂盒扩增(takara公司),引物序列为rd-799f、rd-1193r。
1.4使用agencourtampurexp(beckmancoulter公司,codeno.a63880)对一轮扩增产物进行磁珠纯化,利用上述剪切体系进行体外剪切纯化的第一轮扩增(60ng)。
1.5取1.4中cas9/grna(grnaid为mito1048具体序列见表1)处理的样品3μl为模板,利用p5-index-rd/p7-index-rd引物对不同的处理样品加入不同的index序列(表7)进行扩增,进行第二轮扩增,扩增程序见(表6)。
1.62%琼脂糖凝胶电泳检测cas9/grna剪切去除水稻rrna效果,如图6所示。
1.7将以土壤微生物dna为模板的样品,cas9/grna处理组和对照组,经过pcr第二轮扩增的样品进行磁珠纯化,将文库进行定量均一化处理送样测序。
1.8利用illuminahiseq2500pe250平台进行测序(诺禾致源公司)。
2.对土壤微生物样品16s-seq测序结果进行分析
2.1利用qiime(qiimev1.9.1,http://qiime.org/)对测序数据进行拼接、过滤、去嵌合体序列。
2.2利用uparse软件(uparsev7.0.1001,http://drive5.com/uparse/)对所有样品的全部effectivetags进行聚类,默认以97%的一致性(identity)将序列聚类成为otus,且每个otu的有效序列数大于10。结果统计如表10所示:
表10.样品effectivetags聚类分析
2.3基于otus的cas9/grna处理组和未经cas9/grna处理对照组venn图结果分析(附图7,其中soil#1、soil#2、soil#3代表利用土壤微生物dna为模板构建的扩增子文库的三个生物学重复组)可知,超过80%的otus为这两组共有,仅有低丰度的otus在这两组中有差别。
2.4基于otu出现频率的相关性分析(附图8soil#1、soil#2、soil#3代表利用土壤微生物dna为模板构建的扩增子文库的三个生物学重复组.横坐标代表未利用cas9/grna处理的对照组测序结果的otu出现频率的对数值。纵坐标代表利用cas9/grna处理组测序结果的otu出现频率的对数值,r代表相关性系数)可知,cas9/grna处理组和未经cas9/grna处理对照组otu出现频率具有较高的相关性。
2.5进行样品复杂度分析(alphadiversity).对cas9/grna处理组和未经cas9/grna处理对照组的alphadiversity指数中的simpson指数和shannon指数进行分析(表11),指数越大,说明群落多样性越高。
表11样品复杂度分析
2.6进行多样品比较分析(betadiversity)。通过对cas9/grna处理组和未经cas9/grna处理对照组的测序结果进行upgma分析,结果如图9所示:soil#1、soil#2、soil#3代表利用土壤微生物dna为模板构建的扩增子文库的三个生物学重复组;左侧是不同处理测序样品upgma聚类树结构,右侧的是各样品在门水平上的物种相对丰度分布图,可以直观的看出cas9/grna的处理不会对测序结果产生偏好性影响。
通过上述对cas9/grna处理组和未经cas9/grna处理的对照组的测序结果分析可知,这种基于cas9/grna处理的改进的16s-seq的方法不会对测序结果产生偏好性影响。
实施例4
利用改进的16s-seq的方法分析了水稻根系样品
1.利用cas9/grna处理,构建水稻根系样品16s-seq测序文库1.1取种植在温室42天的mh63水稻根系,置于pbs-s缓冲溶液中,涡旋15s,清洗三次,12000g离心去除沉淀,收集根系,利用灭菌处理研磨钵直接研磨根系,利用fastdnaspinkitforsoil(mb公司)试剂盒,提取水稻根系微生物dna。
1.2利用改进的16s-seq的方法构建水稻根系样品扩增子文库(附图1)。
1.3第一轮扩增(程序见表4)利用16srdnabacterialindentificationpcrkit试剂盒扩增(takara公司),引物序列为rd-799f、rd-1193r。
1.4使用agencourtampurexp(beckmancoulter公司,codeno.a63880)对一轮扩增产物进行磁珠纯化,利用上述剪切体系进行体外剪切纯化的第一轮扩增产物60ng。
1.5取1.4中cas9/grna(grnaid为mito1048具体序列见表1)处理的样品3μl为模板,利用p5-index-rd/p7-index-rd引物对不同的样品组加入不同的index序列(表9)进行第二轮扩增,(扩增程序见表6)。
1.62%琼脂糖凝胶电泳检测cas9/grna剪切去除水稻rrna效果,如图10所示:root#1、root#2、root#3分别代表以提取的水稻根系微生物dna为模板构建的扩增子文库的三个生物学重复组.a图代表利用改进的16s-seq的方法构建扩增子文库的pcr1,b图代表利用cas9/grna处理的pcr1产物,进行pcr2,由于水稻的rrna扩增子序列比微生物rrna序列大90bp,从b图可以看出这种改进的16s-seq的方法在构建水稻根系测序文库时能有效去除大部分的水稻rrna扩增子序列,并能有效富集微生物的rrna扩增子序列。
1.7将以水稻根系微生物dna为模板的样品,cas9/grna处理组和对照组,经过pcr第二轮扩增的样品进行磁珠纯化,将文库进行定量均一化处理送样测序。
2.对水稻根系样品16s-seq测序结果进行分析
2.1对所有样品的全部effectivetags进行聚类,默认以97%的一致性(identity)将序列聚类成为otus,且每个otu的有效序列数大于10。结果统计见表12:
表12样品effectivetags进行聚类分析
2.2对利用改进的16s-seq(cas9/grna处理)的根系样品测序结果,与对照组标准的illumina16s-seq(未经cas9/grna处理)的根系样品测序结果,基于otus的venn图结果分析(图11中root#1、root#2、root#3分别代表以提取的水稻根系微生物dna为模板构建的扩增子文库的三个生物学重复组)可知经过改进的16s-seq的结果能检测到标准的illumina16s-seq检测到的所有otus,并且前者检测到otus是后者的1.5到2倍左右。
2.3对cas9/grna处理组和未经cas9/grna处理对照组共有的otu出现频率进行相关性分析如图12所示:root#1、root#2、root#3分别代表以提取的水稻根系微生物dna为模板构建的扩增子文库的三个生物学重复组.横坐标代表未利用cas9/grna处理的对照组测序结果的otu出现频率的对数值。纵坐标代表利用cas9/grna处理组测序结果的otu出现频率的对数值(r代表相关性系数),由图可知cas9/grna处理组和未经cas9/grna处理对照组共有的otu出现频率具有较高的相关性。
2.4进行样品复杂度(alphadiversity)的稀释曲线分析如图13所示:root#1、root#2、root#3分别代表以提取的水稻根系微生物dna为模板构建的扩增子文库的三个生物学重复组.稀释曲线可直接反映测序数据量的合理性,并间接反映样品中物种的丰富程度,当曲线趋向平坦时,说明测序数据量渐进合理,更多的数据量只会产生少量新的物种(otus)。横坐标为从样品中随机抽取的测序条数,纵坐标为基于该测序条数能构建的otu数量,用来反映测序深度情况.从图中可看出cas9/grna的处理能有效增加测序样品中检测到的otu的数量。稀释曲线是常见的描述组内样品多样性的曲线,是从样品中随机抽取一定测序量的数据,统计它们所代表物种数目(即otus数目),以抽取的测序数据量与对应的物种数来构建曲线。
2.5对测序结果进行upgma聚类分析,如图14所示:root#1、root#2、root#3分别代表以提取的水稻根系微生物dna为模板构建的扩增子文库的三个生物学重复组,左侧是不同处理测序样品upgma聚类树结构,右侧的是各样品在门水平上的物种相对丰度分布图。
通过上述对测序结果进行分析,这种改进的16s-seq的方法能在不引入偏好的情况下明显富集微生物rrna,并且和传统的16s-seq方法相比在相同的测序深度下明显增加检测到的otu的数量。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
序列表
<110>华中农业大学
<120>一种改进的16s-seq方法及其应用
<160>59
<170>siposequencelisting1.0
<210>1
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>1
agttgttgttcccctcccaa20
<210>2
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>2
tccttttgctcctcagccta20
<210>3
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>3
tcagacgcgagcccctcctt20
<210>4
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>4
ccagctactgatcatcgcct20
<210>5
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>5
ttgccccccccgtcttaccg20
<210>6
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>6
tctgtctcactcaagtgaat20
<210>7
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>7
ggcgttccttcgtagatcta20
<210>8
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>8
cagagagctgccttcgcttt20
<210>9
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>9
ttcgcaccccagcgtcggta20
<210>10
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>10
gggcgaacactcatcgttta20
<210>11
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>11
cccctgatccgcgtagacca20
<210>12
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>12
gttgttcatatgtcaagggc20
<210>13
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>13
agtaccatcccgttaaggac20
<210>14
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>14
aagtcagtaccatcccgtta20
<210>15
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>15
acgtaccacaatttctcctt20
<210>16
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>16
ggagaaattgtggtacgtag20
<210>17
<211>76
<212>dna
<213>人工序列(artificialsequence)
<400>17
gttttagagctagaaatagcaagttaaaataaggctagtccgttatcaacttgaaaaagt60
ggcaccgagtcggtgc76
<210>18
<211>52
<212>dna
<213>人工序列(artificialsequence)
<400>18
tcgtcggcagcgtcagatgtgtataagagacagaacmggattagataccckg52
<210>19
<211>53
<212>dna
<213>人工序列(artificialsequence)
<400>19
gtctcgtgggctcggagatgtgtataagagacagcgtcatccmcaccttcctc53
<210>20
<211>29
<212>dna
<213>人工序列(artificialsequence)
<400>20
aatgatacggcgaccaccgagatctacac29
<210>21
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>21
caagcagaagacggcatacgagat24
<210>22
<211>49
<212>dna
<213>人工序列(artificialsequence)
<400>22
ggcggtaccaagctaatacgactcactataggggagaccgaggtctcgg49
<210>23
<211>33
<212>dna
<213>人工序列(artificialsequence)
<400>23
attggatcctttaaaagcaccgactcggtgcca33
<210>24
<211>19
<212>dna
<213>人工序列(artificialsequence)
<400>24
ggtaacgccagggttttcc19
<210>25
<211>16
<212>dna
<213>人工序列(artificialsequence)
<400>25
aaaagcaccgactcgg16
<210>26
<211>18
<212>dna
<213>人工序列(artificialsequence)
<400>26
caggaaacagctatgacc18
<210>27
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>27
taggagttgttgttcccctcccaa24
<210>28
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>28
aaacttgggaggggaacaacaact24
<210>29
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>29
taggtccttttgctcctcagccta24
<210>30
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>30
aaactaggctgaggagcaaaagga24
<210>31
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>31
taggtcagacgcgagcccctcctt24
<210>32
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>32
aaacaaggaggggctcgcgtctga24
<210>33
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>33
taggccagctactgatcatcgcct24
<210>34
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>34
aaacaggcgatgatcagtagctgg24
<210>35
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>35
taggttgccccccccgtcttaccg24
<210>36
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>36
aaaccggtaagacggggggggcaa24
<210>37
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>37
taggtctgtctcactcaagtgaat24
<210>38
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>38
aaacattcacttgagtgagacaga24
<210>39
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>39
taggggcgttccttcgtagatcta24
<210>40
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>40
aaactagatctacgaaggaacgcc24
<210>41
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>41
taggcagagagctgccttcgcttt24
<210>42
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>42
aaacaaagcgaaggcagctctctg24
<210>43
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>43
taggttcgcaccccagcgtcggta24
<210>44
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>44
aaactaccgacgctggggtgcgaa24
<210>45
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>45
tagggggcgaacactcatcgttta24
<210>46
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>46
aaactaaacgatgagtgttcgccc24
<210>47
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>47
taggcccctgatccgcgtagacca24
<210>48
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>48
aaactggtctacgcggatcagggg24
<210>49
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>49
tagggttgttcatatgtcaagggc24
<210>50
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>50
aaacgcccttgacatatgaacaac24
<210>51
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>51
taggagtaccatcccgttaaggac24
<210>52
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>52
aaacgtccttaacgggatggtact24
<210>53
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>53
taggaagtcagtaccatcccgtta24
<210>54
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>54
aaactaacgggatggtactgactt24
<210>55
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>55
taggacgtaccacaatttctcctt24
<210>56
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>56
aaacaaggagaaattgtggtacgt24
<210>57
<211>22
<212>dna
<213>人工序列(artificialsequence)
<400>57
taggagaaattgtggtacgtag22
<210>58
<211>22
<212>dna
<213>人工序列(artificialsequence)
<400>58
aaacctacgtaccacaatttct22
<210>59
<211>2806
<212>dna
<213>人工序列(artificialsequence)
<400>59
tcgcgcgtttcggtgatgacggtgaaaacctctgacacatgcagctcccggagacggtca60
cagcttgtctgtaagcggatgccgggagcagacaagcccgtcagggcgcgtcagcgggtg120
ttggcgggtgtcggggctggcttaactatgcggcatcagagcagattgtactgagagtgc180
accatatgcggtgtgaaataccgcacagatgcgtaaggagaaaataccgcatcaggcgcc240
attcgccattcaggctgcgcaactgttgggaagggcgatcggtgcgggcctcttcgctat300
tacgccagctggcgaaagggggatgtgctgcaaggcgattaagttgggtaacgccagggt360
tttcccagtcacgacgttgtaaaacgacggccagtgaattcgagctcggtaccaagctaa420
tacgactcactataggggagaccgaggtctcgggttttagagctagaaatagcaagttaa480
aataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgcttttaaaggat540
cctctagagtcgacctgcaggcatgcaagcttggcgtaatcatggtcatagctgtttcct600
gtgtgaaattgttatccgctcacaattccacacaacatacgagccggaagcataaagtgt660
aaagcctggggtgcctaatgagtgagctaactcacattaattgcgttgcgctcactgccc720
gctttccagtcgggaaacctgtcgtgccagctgcattaatgaatcggccaacgcgcgggg780
agaggcggtttgcgtattgggcgctcttccgcttcctcgctcactgactcgctgcgctcg840
gtcgttcggctgcggcgagcggtatcagctcactcaaaggcggtaatacggttatccaca900
gaatcaggggataacgcaggaaagaacatgtgagcaaaaggccagcaaaaggccaggaac960
cgtaaaaaggccgcgttgctggcgtttttccataggctccgcccccctgacgagcatcac1020
aaaaatcgacgctcaagtcagaggtggcgaaacccgacaggactataaagataccaggcg1080
tttccccctggaagctccctcgtgcgctctcctgttccgaccctgccgcttaccggatac1140
ctgtccgcctttctcccttcgggaagcgtggcgctttctcatagctcacgctgtaggtat1200
ctcagttcggtgtaggtcgttcgctccaagctgggctgtgtgcacgaaccccccgttcag1260
cccgaccgctgcgccttatccggtaactatcgtcttgagtccaacccggtaagacacgac1320
ttatcgccactggcagcagccactggtaacaggattagcagagcgaggtatgtaggcggt1380
gctacagagttcttgaagtggtggcctaactacggctacactagaaggacagtatttggt1440
atctgcgctctgctgaagccagttaccttcggaaaaagagttggtagctcttgatccggc1500
aaacaaaccaccgctggtagcggtggtttttttgtttgcaagcagcagattacgcgcaga1560
aaaaaaggatctcaagaagatcctttgatcttttctacggggtctgacgctcagtggaac1620
gaaaactcacgttaagggattttggtcatgagattatcaaaaaggatcttcacctagatc1680
cttttaaattaaaaatgaagttttaaatcaatctaaagtatatatgagtaaacttggtct1740
gacagttaccaatgcttaatcagtgaggcacctatctcagcgatctgtctatttcgttca1800
tccatagttgcctgactccccgtcgtgtagataactacgatacgggagggcttaccatct1860
ggccccagtgctgcaatgataccgcgagtgccacgctcaccggctccagatttatcagca1920
ataaaccagccagccggaagggccgagcgcagaagtggtcctgcaactttatccgcctcc1980
atccagtctattaattgttgccgggaagctagagtaagtagttcgccagttaatagtttg2040
cgcaacgttgttgccattgctacaggcatcgtggtgtcacgctcgtcgtttggtatggct2100
tcattcagctccggttcccaacgatcaaggcgagttacatgatcccccatgttgtgcaaa2160
aaagcggttagctccttcggtcctccgatcgttgtcagaagtaagttggccgcagtgtta2220
tcactcatggttatggcagcactgcataattctcttactgtcatgccatccgtaagatgc2280
ttttctgtgactggtgagtactcaaccaagtcattctgagaatagtgtatgcggcgaccg2340
agttgctcttgcccggcgtcaatacgggataataccgcgccacatagcagaactttaaaa2400
gtgctcatcattggaaaacgttcttcggggcgaaaactctcaaggatcttaccgctgttg2460
agatccagttcgatgtaacccactcgtgcacccaactgatcttcagcatcttttactttc2520
accagcgtttctgggtgagcaaaaacaggaaggcaaaatgccgcaaaaaagggaataagg2580
gcgacacggaaatgttgaatactcatactcttcctttttcaatattattgaagcatttat2640
cagggttattgtctcatgagcggatacatatttgaatgtatttagaaaaataaacaaata2700
ggggttccgcgcacatttccccgaaaagtgccacctgacgtctaagaaaccattattatc2760
atgacattaacctataaaaataggcgtatcacgaggccctttcgtc2806
- 还没有人留言评论。精彩留言会获得点赞!