舒巴坦酸的合成方法与流程
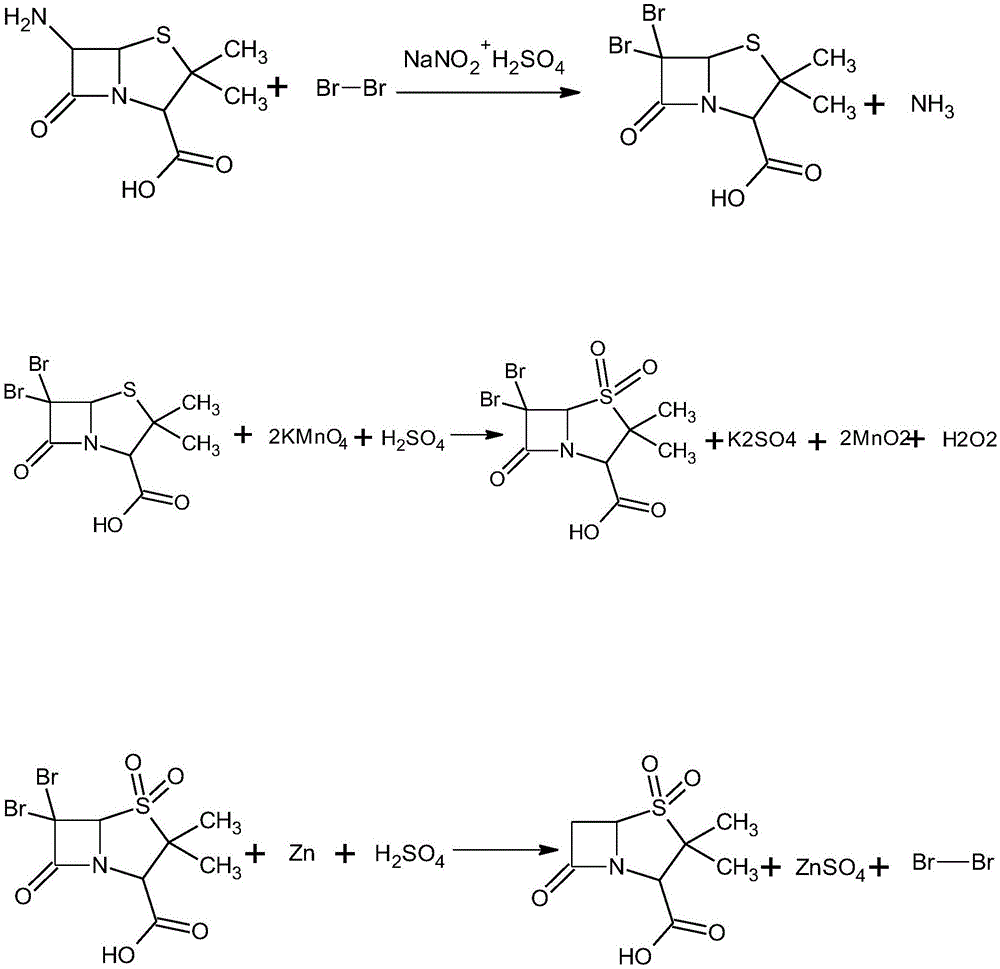
本发明涉及药物合成领域,更具体地说,它涉及一种舒巴坦酸的合成方法。
背景技术:
舒巴坦酸是一种半合成的广谱β-内酰胺酶抑制剂,它是一种竞争性不可逆的β-内酰胺酶抑制剂,与β-内酰胺类抗生素合用,有很好的协同作用,近年来在医药上已得到广泛的应用。舒巴坦酸的结构式如下所示。
舒巴坦酸为类白色或淡黄色结晶性粉末,易溶于水、醇、酯,难溶于醚。舒巴坦酸是头孢类抗菌药物和青霉素抗菌药物的首选酶抑制剂,属于非典型β-内酰胺类药物。由于头孢类和青霉素类药物的长期大量使用,使得β-内酰胺酶产生了抗生素的耐药性,而舒巴坦酸与头孢类和青霉素类药物配合使用后,通过抑制β-内酰胺酶的产生,解决了抗生素耐药性的难题,是国内外使用最广泛的β-内酰胺酶抑制剂。
舒巴坦酸传统的合成工艺通常是以6-氨基青霉烷酸为起始原料,经与亚硝酸钠在酸性条件下进行重氮化反应,然后与溴素进行双溴化反应,再经过高锰酸钾氧化,最后用金属粉末锌粉、镁粉或催化氢化还原制得所需化合物舒巴坦酸。但是,利用传统工艺制备舒巴坦酸的反应收率普遍约为70%,收率较低,仍有改进的空间。
技术实现要素:
针对现有技术存在的不足,本发明的目的在于提供一种舒巴坦酸的合成方法,具有提高反应收率的优点。
为实现上述目的,本发明提供了如下技术方案:
一种舒巴坦酸的合成方法,包括以下步骤:
s1、重氮化及溴化反应:在6-氨基青霉烷酸中加入溴素、稀硫酸溶液以及亚硝酸钠固体,所述稀硫酸溶液的浓度为20%-25%,所述6-氨基青霉烷酸与溴素的摩尔比为1:1.5-2,所述6-氨基青霉烷酸与稀硫酸的摩尔比为1:1-2,发生重氮化反应以及溴化反应,得到第一中间体;
s2、氧化反应:向第一中间体中滴加高锰酸钾以及稀硫酸溶液,所述稀硫酸溶液的浓度为20%-25%,所述高锰酸钾与稀硫酸的摩尔比为2:1-1.5,发生氧化反应,得到第二中间体;
s3、氢化反应:向第二中间体中滴加锌粉以及稀硫酸溶液,发生氢化反应,得到产物舒巴坦酸。
采用上述技术方案,通过采用20%-25%的稀硫酸溶液进行反应,有利于提高重氮化及溴化反应和氧化反应的活性,使得反应物反应得更加彻底,从而有利于提高重氮化及溴化反应和氧化反应的收率,进而使得反应的总收率提高;同时,通过6-氨基青霉烷酸与溴素的摩尔比为1:1.5-2以及6-氨基青霉烷酸与稀硫酸的摩尔比为1:1-2,有利于减少副反应的发生,使得反应物反应得更加彻底,从而使得重氮化及溴化反应的转化率提高,使得重氮化及溴化反应的收率提高,进而有利于提高反应的总收率;通过加入的高锰酸钾与稀硫酸的摩尔比为2:1-1.5,有利于减少副反应的产生,使得反应物反应得更加彻底,有利于提高氧化反应的反应转化率,使得氧化反应的收率提高,进而使得反应的总收率提高。
本发明进一步设置为:还包括以下步骤:
s4、萃取、干燥:过滤反应后的反应产物,取滤液静置分层,萃取水相后合并至有机相中,脱色,离心脱水,最后放入烘干箱中干燥,得成品。
采用上述技术方案,通过对产物的水相进行萃取再合并至有机相中的操作,使得溶解于水相中的产物被重新萃取至有机相中得以收集,从而有利于减少产物的损失,使得反应的总收率提高;通过对产物进行离心脱水以及干燥的操作,有利于除去产物中的水分,使得产物的纯度提高。
本发明进一步设置为:所述步骤s4中,在脱色后,离心脱水之前,先对产物进行减压蒸馏。
采用上述技术方案,通过在离心脱水之前,先对产物进行减压蒸馏的操作,有利于提高产物的纯度,另外,在减压条件下进行蒸馏,有利于增强提纯的效果,使得产物的纯度更高。
本发明进一步设置为:所述步骤s1的反应温度控制在25℃-30℃。
采用上述技术方案,通过步骤s1的反应温度控制在25℃-30℃,有利于重氮化及溴化反应的进行,使得反应物反应得更加彻底,有利于提高重氮化及溴化反应的转化率,使得反应收率提高,进而有利于提高反应的总收率。
本发明进一步设置为:所述步骤s2的反应温度控制在5℃-12℃。
采用上述技术方案,通过步骤s2的反应温度控制在5℃-12℃,有利于氧化反应的进行,有利于反应物尽可能地转化为产物,使得氧化反应的转化率提高,有利于提高氧化反应的收率,进而有利于提高反应的总收率。
本发明进一步设置为:所述步骤s3的反应温度控制在10℃-12℃。
采用上述技术方案,通过步骤s3的反应温度控制在10℃-12℃,有利于氢化反应的进行,使得反应物反应得更加彻底,有利于提高氢化反应的转化率,从而使得氢化反应的收率提高,进而有利于提高反应的总收率。
本发明进一步设置为:所述步骤s1中,先将溴素、稀硫酸溶液以及亚硝酸钠混合均匀形成反应液后,再加入至6-氨基青霉烷酸中进行反应。
采用上述技术方案,通过先将溴素、稀硫酸溶液以及亚硝酸钠混合均匀形成反应液后,再加入至6-氨基青霉烷酸中,有利于减少副反应的产生,有利于6-氨基青霉烷酸尽可能地转化为第一中间体,使得重氮化及溴化反应的转化率提高,从而有利于提高重氮化及溴化反应的收率,进而使得反应的总收率提高。
本发明进一步设置为:所述步骤s2中,先将稀硫酸溶液以及高锰酸钾溶液混合均匀以形成氧化液后,再将氧化液滴加至第一中间体中反应。
采用上述技术方案,通过先将稀硫酸溶液以及高锰酸钾溶液混合均匀形成氧化液后,再将氧化液滴加至第一中间体中反应,有利于第一中间体更彻底地转化为第二中间体,使得氧化反应的转化率提高,从而使得氧化反应的收率提高,进而有利于提高反应的总收率。
本发明进一步设置为:所述步骤s2中,在进行氧化反应之前,先调节第一中间体的ph至6.8-7.1。
采用上述技术方案,通过在进行氧化反应之前,先调节第一中间体的ph至6.8-7.1,有利于第一中间体更好地与高锰酸钾以及稀硫酸反应,使得氧化反应的转化率提高,从而使得氧化反应的收率提高,进而有利于提高反应的总收率。
本发明进一步设置为:所述步骤s4中,减压蒸馏操作的压力条件控制为-0.09mpa。
采用上述技术方案,通过减压蒸馏操作的压力条件控制为-0.09mpa,有利于提高蒸馏的效率和蒸馏的效果,使得蒸馏时间缩短,同时有利于提高蒸馏所得产物的纯度。
综上所述,本发明具有以下有益效果:
1.通过采用20%-25%的稀硫酸溶液进行反应,有利于提高重氮化及溴化反应和氧化反应的活性,使得反应物反应得更加彻底,从而有利于提高重氮化及溴化反应和氧化反应的收率,进而使得反应的总收率提高;
2.通过6-氨基青霉烷酸与溴素的摩尔比为1:1.5-2以及6-氨基青霉烷酸与稀硫酸的摩尔比为1:1-2,有利于减少副反应的发生,使得反应物反应得更加彻底,从而使得重氮化及溴化反应的收率提高,进而有利于提高反应的总收率;
3.通过加入的高锰酸钾与稀硫酸的摩尔比为2:1-1.5,有利于减少副反应的产生,使得反应物反应得更加彻底,有利于提高氧化反应的收率,进而使得反应的总收率提高;
4.通过对产物的水相进行萃取再合并至有机相中的操作,使得溶解于水相中的产物被重新萃取至有机相中得以收集,从而有利于减少产物的损失,使得反应的总收率提高;
5.通过对产物进行离心脱水以及干燥的操作,有利于除去产物中的水分,使得产物的纯度提高。
附图说明
图1为本发明中舒巴坦酸的合成路径;
图2为舒巴坦酸的液相色谱谱图。
具体实施方式
以下结合附图及实施例,对本发明作进一步详细说明。
实施例1
一种舒巴坦酸的合成方法,包括以下步骤:
s1、重氮化及溴化反应,具体如下:
在1号反应釜中加入乙酸乙酯843g,降温至0℃,以50r/min的转速进行搅拌,边搅拌边滴加溴素240g(1.5mol)以及纯水420g,控制温度为3℃,再滴加20%的稀硫酸490g(1mol)。
控制温度为4℃,以250r/min的转速进行搅拌,边搅拌边加入亚硝酸钠固体103.5g,亚硝酸钠固体分5次加入,加入时间为15min。
将216g(1mol)的6-氨基青霉烷酸用真空上料机从投料口加入1号反应釜中,控制温度为25℃,保温60min,反应结束。
再向1号反应釜中滴加亚硫酸氢钠溶液(750g水+375g亚硫酸氢钠),控制温度为10℃,使物料颜色褪色。物料为淡黄色,用淀粉-碘化钾指示剂检测,当滴加至淀粉-碘化钾指示剂5秒内不变蓝时,停止滴加。
静置分层,将水相转移至2号萃取釜中,采用450g的乙酸乙酯分三次萃取,每次用150g。萃取完成后再将2号萃取釜中的有机相转移至1号反应釜中,并向1号反应釜中滴加175g纯水进行洗涤,洗涤完成后用稀碱液(625g水+187g氢氧化钠)调节ph至6.8,并控制温度为5℃,得到第一中间体。
重氮化及溴化反应的反应式如下所示。
s2、氧化反应,具体如下:
配制氧化液:在真空条件下往3号反应釜中加入纯水900g、20%的稀硫酸溶液490g(1mol),再通过投料口投入高锰酸钾316g(2mol),并搅拌溶解以形成氧化液。
将1号反应釜中的第一中间体冷却至10℃,在10℃的温度条件下向1号反应釜中滴加氧化液,滴加时间为1h。加完在5℃的温度条件下反应30min。
再将1号反应釜中的温度降至2℃,并向1号反应釜中滴加15%的亚硫酸氢钠溶液,直至物料褪色。
静置分层,将水层转移至4号萃取釜中,采用450g的乙酸乙酯分三次萃取,每次用150g。萃取完后再将4号萃取釜中的有机相转移至1号反应釜中,并向1号反应釜中滴加175g的纯水进行洗涤,得到第二中间体。
氧化反应的反应式如下所示。
s3、氢化反应,具体如下:
在5号还原釜中加入纯水1090g,再将1号反应釜中的物料加入5号还原釜中。控制温度为10℃,加入锌粉4g,搅拌5min。
再在真空条件下慢慢抽入20%的稀硫酸溶液531g,然后在0℃的温度条件下慢慢加入183.5g锌粉,加锌粉的时间控制为30min,锌粉加完后,搅拌反应30min,得到产物舒巴坦酸。
氢化反应的反应式如下所示。
s4、萃取并干燥,具体如下:
反应结束后,过滤反应所得的产物舒巴坦酸,取滤液转移至6号还原釜中,静置分层。将水相转移至7号萃取釜中,采用450g的乙酸乙酯分三次萃取,每次150g,萃取完后再将7号萃取釜中的有机相以及5号还原釜中剩余的有机相合并至8号脱色釜中,加入活性炭7.5g,在室温下脱色30min,然后进行过滤,取滤液转移至9号结晶釜中。
控制釜内温度为20℃,控制压力为-0.09mpa,对9号结晶釜中的滤液进行减压蒸馏操作,蒸馏至滤液析出结晶。并注意观察滤液的状态,当蒸馏浓缩溶液逐渐开始发白时,注意观察固体物料的状态,当固体物料比较黏稠,流动性比较顺畅时,停止浓缩蒸馏。
将蒸馏所得的固体物料放入离心机中离心甩干,然后用125g乙酸乙酯泡洗再甩干。
将离心物料投入双锥干燥器中,先室温干燥1h,再打开热水系统,升温至42℃,待温度稳定后,再恒温干燥5h。
关掉热水,再抽真空0.5h,并取样送化验测水分含量,当水分含量不大于0.5%时,即可出料分装,得到成品259g,总收率94%。
实施例2
一种舒巴坦酸的合成方法,包括以下步骤:
s1、重氮化及溴化反应,具体如下:
在1号反应釜中加入乙酸乙酯952g,降温至3℃,以50r/min的转速进行搅拌,边搅拌边滴加溴素272g(1.7mol)以及纯水474g,控制温度为4℃,再滴加23%的稀硫酸639g(1.5mol)。
控制温度为6℃,以250r/min的转速进行搅拌,边搅拌边加入亚硝酸钠固体117g,亚硝酸钠固体分5次加入,加入时间为17min。
将216g(1mol)的6-氨基青霉烷酸用真空上料机从投料口加入1号反应釜中,控制温度为27℃,保温60min,反应结束。
再向1号反应釜中滴加亚硫酸氢钠溶液(750g水+375g亚硫酸氢钠),控制温度为13℃,使物料颜色褪色。物料为淡黄色,用淀粉-碘化钾指示剂检测,当滴加至淀粉-碘化钾指示剂5秒内不变蓝时,停止滴加。
静置分层,将水相转移至2号萃取釜中,采用507g的乙酸乙酯分三次萃取,每次用169g。萃取完成后再将2号萃取釜中的有机相转移至1号反应釜中,并向1号反应釜中滴加198g纯水进行洗涤,洗涤完成后用稀碱液(625g水+187g氢氧化钠)调节ph至6.9,并控制温度为8℃,得到第一中间体。
s2、氧化反应,具体如下:
配制氧化液:在真空条件下往3号反应釜中加入纯水1020g、23%的稀硫酸溶液554g(1.3mol),再通过投料口投入高锰酸钾316g(2mol),并搅拌溶解以形成氧化液。
将1号反应釜中的第一中间体冷却至13℃,在15℃的温度条件下向1号反应釜中滴加氧化液,滴加时间为1h。加完在9℃的温度条件下反应30min。
再将1号反应釜中的温度降至3℃,并向1号反应釜中滴加15%的亚硫酸氢钠溶液,直至物料褪色。
静置分层,将水层转移至4号萃取釜中,采用507g的乙酸乙酯分三次萃取,每次用169g。萃取完后再将4号萃取釜中的有机相转移至1号反应釜中,并向1号反应釜中滴加198g的纯水进行洗涤,得到第二中间体。
s3、氢化反应,具体如下:
在5号还原釜中加入纯水1232g,再将1号反应釜中的物料加入5号还原釜中。控制温度为11℃,加入锌粉4g,搅拌5min。
再在真空条件下慢慢抽入23%的稀硫酸溶液600g,然后在5℃的温度条件下慢慢加入207g锌粉,加锌粉的时间控制为40min,锌粉加完后,搅拌反应30min,得到产物舒巴坦酸。
s4、萃取并干燥,具体如下:
反应结束后,过滤反应所得的产物舒巴坦酸,取滤液转移至6号还原釜中,静置分层。将水相转移至7号萃取釜中,采用507g的乙酸乙酯分三次萃取,每次169g,萃取完后再将7号萃取釜中的有机相以及5号还原釜中剩余的有机相合并至8号脱色釜中,加入活性炭8.5g,在室温下脱色30min,然后进行过滤,取滤液转移至9号结晶釜中。
控制釜内温度为26℃,控制压力为-0.09mpa,对9号结晶釜中的滤液进行减压蒸馏操作,蒸馏至滤液析出结晶。并注意观察滤液的状态,当蒸馏浓缩溶液逐渐开始发白时,注意观察固体物料的状态,当固体物料比较黏稠,流动性比较顺畅时,停止浓缩蒸馏。
将蒸馏所得的固体物料放入离心机中离心甩干,然后用141g乙酸乙酯泡洗再甩干。
将离心物料投入双锥干燥器中,先室温干燥1h,再打开热水系统,升温至43℃,待温度稳定后,再恒温干燥5h。
关掉热水,再抽真空0.5h,并取样送化验测水分含量,当水分含量不大于0.5%时,即可出料分装,得到成品261g,总收率95%。
实施例3
一种舒巴坦酸的合成方法,包括以下步骤:
s1、重氮化及溴化反应,具体如下:
在1号反应釜中加入乙酸乙酯852g,降温至5℃,以50r/min的转速进行搅拌,边搅拌边滴加溴素320g(2mol)以及纯水430g,控制温度为5℃,再滴加25%的稀硫酸784g(2mol)。
控制温度为8℃,以250r/min的转速进行搅拌,边搅拌边加入亚硝酸钠固体113.5g,亚硝酸钠固体分5次加入,加入时间为20min。
将216g(1mol)的6-氨基青霉烷酸用真空上料机从投料口加入1号反应釜中,控制温度为30℃,保温60min,反应结束。
再向1号反应釜中滴加亚硫酸氢钠溶液(750g水+375g亚硫酸氢钠),控制温度为15℃,使物料颜色褪色。物料为淡黄色,用淀粉-碘化钾指示剂检测,当滴加至淀粉-碘化钾指示剂5秒内不变蓝时,停止滴加。
静置分层,将水相转移至2号萃取釜中,采用540g的乙酸乙酯分三次萃取,每次用180g。萃取完成后再将2号萃取釜中的有机相转移至1号反应釜中,并向1号反应釜中滴加186g纯水进行洗涤,洗涤完成后用稀碱液(625g水+187g氢氧化钠)调节ph至7.1,并控制温度为12℃,得到第一中间体。
s2、氧化反应,具体如下:
配制氧化液:在真空条件下往3号反应釜中加入纯水1000g、25%的稀硫酸溶液588g(1.5mol),再通过投料口投入高锰酸钾316g(2mol),并搅拌溶解以形成氧化液。
将1号反应釜中的第一中间体冷却至15℃,在20℃的温度条件下向1号反应釜中滴加氧化液,滴加时间为1h。加完在12℃的温度条件下反应30min。
再将1号反应釜中的温度降至5℃,并向1号反应釜中滴加15%的亚硫酸氢钠溶液,直至物料褪色。
静置分层,将水层转移至4号萃取釜中,采用540g的乙酸乙酯分三次萃取,每次用180g。萃取完后再将4号萃取釜中的有机相转移至1号反应釜中,并向1号反应釜中滴加210g的纯水进行洗涤,得到第二中间体。
s3、氢化反应,具体如下:
在5号还原釜中加入纯水1300g,再将1号反应釜中的物料加入5号还原釜中。控制温度为12℃,加入锌粉4g,搅拌5min。
再在真空条件下慢慢抽入25%的稀硫酸溶液488g,然后在10℃的温度条件下慢慢加入211g锌粉,加锌粉的时间控制为50min,锌粉加完后,搅拌反应30min,得到产物舒巴坦酸。
s4、萃取并干燥,具体如下:
反应结束后,过滤反应所得的产物舒巴坦酸,取滤液转移至6号还原釜中,静置分层。将水相转移至7号萃取釜中,采用540g的乙酸乙酯分三次萃取,每次180g,萃取完后再将7号萃取釜中的有机相以及5号还原釜中剩余的有机相合并至8号脱色釜中,加入活性炭8g,在室温下脱色30min,然后进行过滤,取滤液转移至9号结晶釜中。
控制釜内温度为32℃,控制压力为-0.09mpa,对9号结晶釜中的滤液进行减压蒸馏操作,蒸馏至滤液析出结晶。并注意观察滤液的状态,当蒸馏浓缩溶液逐渐开始发白时,注意观察固体物料的状态,当固体物料比较黏稠,流动性比较顺畅时,停止浓缩蒸馏。
将蒸馏所得的固体物料放入离心机中离心甩干,然后用150g乙酸乙酯泡洗再甩干。
将离心物料投入双锥干燥器中,先室温干燥1h,再打开热水系统,升温至45℃,待温度稳定后,再恒温干燥5h。
关掉热水,再抽真空0.5h,并取样送化验测水分含量,当水分含量不大于0.5%时,即可出料分装,得到成品260g,总收率94%。
实施例4
一种舒巴坦酸的合成方法,包括以下步骤:
s1、重氮化及溴化反应,具体如下:
在1号反应釜中加入乙酸乙酯845g,降温至4℃,以50r/min的转速进行搅拌,边搅拌边滴加溴素288g(1.8mol)以及纯水431g,控制温度为3℃,再滴加24%的稀硫酸694g(1.7mol)。
控制温度为7℃,以250r/min的转速进行搅拌,边搅拌边加入亚硝酸钠固体112g,亚硝酸钠固体分5次加入,加入时间为18min。
将216g(1mol)的6-氨基青霉烷酸用真空上料机从投料口加入1号反应釜中,控制温度为29℃,保温60min,反应结束。
再向1号反应釜中滴加亚硫酸氢钠溶液(750g水+375g亚硫酸氢钠),控制温度为11℃,使物料颜色褪色。物料为淡黄色,用淀粉-碘化钾指示剂检测,当滴加至淀粉-碘化钾指示剂5秒内不变蓝时,停止滴加。
静置分层,将水相转移至2号萃取釜中,采用456g的乙酸乙酯分三次萃取,每次用152g。萃取完成后再将2号萃取釜中的有机相转移至1号反应釜中,并向1号反应釜中滴加182g纯水进行洗涤,洗涤完成后用稀碱液(625g水+187g氢氧化钠)调节ph至7.0,并控制温度为10℃,得到第一中间体。
s2、氧化反应,具体如下:
配制氧化液:在真空条件下往3号反应釜中加入纯水920g、24%的稀硫酸溶液571.7g(1.4mol),再通过投料口投入高锰酸钾316g(2mol),并搅拌溶解以形成氧化液。
将1号反应釜中的第一中间体冷却至11℃,在19℃的温度条件下向1号反应釜中滴加氧化液,滴加时间为1h。加完在10℃的温度条件下反应30min。
再将1号反应釜中的温度降至4℃,并向1号反应釜中滴加15%的亚硫酸氢钠溶液,直至物料褪色。
静置分层,将水层转移至4号萃取釜中,采用456g的乙酸乙酯分三次萃取,每次用152g。萃取完后再将4号萃取釜中的有机相转移至1号反应釜中,并向1号反应釜中滴加183g的纯水进行洗涤,得到第二中间体。
s3、氢化反应,具体如下:
在5号还原釜中加入纯水1050g,再将1号反应釜中的物料加入5号还原釜中。控制温度为10℃,加入锌粉4g,搅拌5min。
再在真空条件下慢慢抽入24%的稀硫酸溶液553g,然后在8℃的温度条件下慢慢加入186g锌粉,加锌粉的时间控制为47min,锌粉加完后,搅拌反应30min,得到产物舒巴坦酸。
s4、萃取并干燥,具体如下:
反应结束后,过滤反应所得的产物舒巴坦酸,取滤液转移至6号还原釜中,静置分层。将水相转移至7号萃取釜中,采用456g的乙酸乙酯分三次萃取,每次152g,萃取完后再将7号萃取釜中的有机相以及5号还原釜中剩余的有机相合并至8号脱色釜中,加入活性炭9g,在室温下脱色30min,然后进行过滤,取滤液转移至9号结晶釜中。
控制釜内温度为25℃,控制压力为-0.09mpa,对9号结晶釜中的滤液进行减压蒸馏操作,蒸馏至滤液析出结晶。并注意观察滤液的状态,当蒸馏浓缩溶液逐渐开始发白时,注意观察固体物料的状态,当固体物料比较黏稠,流动性比较顺畅时,停止浓缩蒸馏。
将蒸馏所得的固体物料放入离心机中离心甩干,然后用133g乙酸乙酯泡洗再甩干。
将离心物料投入双锥干燥器中,先室温干燥1h,再打开热水系统,升温至43℃,待温度稳定后,再恒温干燥5h。
关掉热水,再抽真空0.5h,并取样送化验测水分含量,当水分含量不大于0.5%时,即可出料分装,得到成品257g,总收率93%。
比较例1
本实施例参见公开号为cn102952147b,专利名称为一种舒巴坦酸的合成方法的专利文件中的合成方法。具体合成方法如下:
s1:在1500l反应锅中加入1000kg乙酸乙酯,然后搅拌冷至0℃,吸入溴素150kg,滴加硫酸水溶液(硫酸取43.2kg+水200kg),控制温度不超过5℃,冷至4℃,分数次加入57.6kg亚硝酸钠,控制温度不超过5℃,保温30分钟,然后于10℃分次慢慢加入6-氨基青霉烷酸90kg,加完再于10℃反应0.4小时,保温结束后在14℃滴加10%亚硫酸氢钠水溶液至溴色消失,反应液呈浅黄色,待无黄色气体逸出即可停止搅拌,静置分层,下层水层用乙酸乙酯提取二次(400kg×2),提取后的水层回收溴素,合并乙酸乙酯层至中和锅搅拌冷却,用氢氧化钠液调ph为7,静止分层,乙酸乙酯用水提取两次(200kg×2),溴化水层抽入氧化锅中,乙酸乙酯下批套用。
s2:将氧化反应锅的溴化水层冷至10℃以下,滴加先配好的高锰酸钾酸性水溶液,温度控制在10℃,加完后再于10℃反应30分钟,加入500kg乙酸乙酯降温至5℃,用30%盐酸调ph至1.25,于5℃滴加10%亚硫酸氢钠水溶液,滴至高锰酸钾色消失,而反应液呈浅黄色为终点复测ph,如ph值高于1.25,再用30%盐酸调ph至1.25,加入50kg精盐搅拌20分钟,使其饱和(如不饱和可补加些),静置分层,水层用乙酸乙酯提取两次(250kg×2),水层抽入处理水缸,合并乙酸乙酯层。得到第一中间产物。
s3:将第一中间产物抽到3000l还原锅中,冷却到0℃以下开始加入乙酸乙酯(上述步骤中所回收)和水。打开放空阀门,开搅拌降温。待内温达至0℃开始分次加入镁粉,并同时滴加硫酸保持ph不超过5,待温度不明显上升且ph值下降时,保温15分钟,然后冷却10℃,调ph至1.25,过滤分层,水层用乙酸乙酯(500kg)提取一次,水层抽处理池处理,合并乙酸乙酯,并抽入一批待套用的母液,用100kg饱和盐水洗涤一次,分净后加活性炭脱色,脱色完成后过滤,然后抽滤到蒸馏锅蒸馏到液料基本干时,将结晶物放至离心机甩滤,甩干后用约50kg的醋酸乙酯淋洗,甩干后出料。并在真空度≤-0.085mpa,水浴温度在28-35℃的双锥中干燥3小时,烘干。总收率77%。
比较例2
一种舒巴坦酸的合成方法,包括以下步骤:
s1、重氮化及溴化反应,具体如下:
在1号反应釜中加入乙酸乙酯843g,降温至0℃,以50r/min的转速进行搅拌,边搅拌边滴加溴素240g(1.5mol)以及纯水420g,控制温度为3℃,再滴加30%的稀硫酸326.7g(1mol)。
控制温度为4℃,以250r/min的转速进行搅拌,边搅拌边加入亚硝酸钠固体103.5g,亚硝酸钠固体分5次加入,加入时间为15min。
将216g(1mol)的6-氨基青霉烷酸用真空上料机从投料口加入1号反应釜中,控制温度为25℃,保温60min,反应结束。
再向1号反应釜中滴加亚硫酸氢钠溶液(750g水+375g亚硫酸氢钠),控制温度为10℃,使物料颜色褪色。物料为淡黄色,用淀粉-碘化钾指示剂检测,当滴加至淀粉-碘化钾指示剂5秒内不变蓝时,停止滴加。
静置分层,将水相转移至2号萃取釜中,采用450g的乙酸乙酯分三次萃取,每次用150g。萃取完成后再将2号萃取釜中的有机相转移至1号反应釜中,并向1号反应釜中滴加175g纯水进行洗涤,洗涤完成后用稀碱液(625g水+187g氢氧化钠)调节ph至6.8,并控制温度为5℃,得到第一中间体。
s2、氧化反应,具体如下:
配制氧化液:在真空条件下往3号反应釜中加入纯水900g、30%的稀硫酸溶液326.7g(1mol),再通过投料口投入高锰酸钾316g(2mol),并搅拌溶解以形成氧化液。
将1号反应釜中的第一中间体冷却至10℃,在10℃的温度条件下向1号反应釜中滴加氧化液,滴加时间为1h。加完在5℃的温度条件下反应30min。
再将1号反应釜中的温度降至2℃,并向1号反应釜中滴加15%的亚硫酸氢钠溶液,直至物料褪色。
静置分层,将水层转移至4号萃取釜中,采用450g的乙酸乙酯分三次萃取,每次用150g。萃取完后再将4号萃取釜中的有机相转移至1号反应釜中,并向1号反应釜中滴加175g的纯水进行洗涤,得到第二中间体。
s3、氢化反应,具体如下:
在5号还原釜中加入纯水1090g,再将1号反应釜中的物料加入5号还原釜中。控制温度为10℃,加入锌粉4g,搅拌5min。
再在真空条件下慢慢抽入30%的稀硫酸溶液354g,然后在0℃的温度条件下慢慢加入183.5g锌粉,加锌粉的时间控制为30min,锌粉加完后,搅拌反应30min,得到产物舒巴坦酸。
s4、萃取并干燥,具体如下:
反应结束后,过滤反应所得的产物舒巴坦酸,取滤液转移至6号还原釜中,静置分层。将水相转移至7号萃取釜中,采用450g的乙酸乙酯分三次萃取,每次150g,萃取完后再将7号萃取釜中的有机相以及5号还原釜中剩余的有机相合并至8号脱色釜中,加入活性炭7.5g,在室温下脱色30min,然后进行过滤,取滤液转移至9号结晶釜中。
控制釜内温度为20℃,控制压力为-0.09mpa,对9号结晶釜中的滤液进行减压蒸馏操作,蒸馏至滤液析出结晶。并注意观察滤液的状态,当蒸馏浓缩溶液逐渐开始发白时,注意观察固体物料的状态,当固体物料比较黏稠,流动性比较顺畅时,停止浓缩蒸馏。
将蒸馏所得的固体物料放入离心机中离心甩干,然后用125g乙酸乙酯泡洗再甩干。
将离心物料投入双锥干燥器中,先室温干燥1h,再打开热水系统,升温至42℃,待温度稳定后,再恒温干燥5h。
关掉热水,再抽真空0.5h,并取样送化验测水分含量,当水分含量不大于0.5%时,即可出料分装,得到成品215g,总收率78%。
比较例3
一种舒巴坦酸的合成方法,包括以下步骤:
s1、重氮化及溴化反应,具体如下:
在1号反应釜中加入乙酸乙酯952g,降温至3℃,以50r/min的转速进行搅拌,边搅拌边滴加溴素192g(1.2mol)以及纯水474g,控制温度为4℃,再滴加23%的稀硫酸213g(0.5mol)。
控制温度为6℃,以250r/min的转速进行搅拌,边搅拌边加入亚硝酸钠固体117g,亚硝酸钠固体分5次加入,加入时间为17min。
将216g(1mol)的6-氨基青霉烷酸用真空上料机从投料口加入1号反应釜中,控制温度为27℃,保温60min,反应结束。
再向1号反应釜中滴加亚硫酸氢钠溶液(750g水+375g亚硫酸氢钠),控制温度为13℃,使物料颜色褪色。物料为淡黄色,用淀粉-碘化钾指示剂检测,当滴加至淀粉-碘化钾指示剂5秒内不变蓝时,停止滴加。
静置分层,将水相转移至2号萃取釜中,采用507g的乙酸乙酯分三次萃取,每次用169g。萃取完成后再将2号萃取釜中的有机相转移至1号反应釜中,并向1号反应釜中滴加198g纯水进行洗涤,洗涤完成后用稀碱液(625g水+187g氢氧化钠)调节ph至6.9,并控制温度为8℃,得到第一中间体。
s2、氧化反应,具体如下:
配制氧化液:在真空条件下往3号反应釜中加入纯水1020g、23%的稀硫酸溶液213g(0.5mol),再通过投料口投入高锰酸钾316g(2mol),并搅拌溶解以形成氧化液。
将1号反应釜中的第一中间体冷却至13℃,在15℃的温度条件下向1号反应釜中滴加氧化液,滴加时间为1h。加完在9℃的温度条件下反应30min。
再将1号反应釜中的温度降至3℃,并向1号反应釜中滴加15%的亚硫酸氢钠溶液,直至物料褪色。
静置分层,将水层转移至4号萃取釜中,采用507g的乙酸乙酯分三次萃取,每次用169g。萃取完后再将4号萃取釜中的有机相转移至1号反应釜中,并向1号反应釜中滴加198g的纯水进行洗涤,得到第二中间体。
s3、氢化反应,具体如下:
在5号还原釜中加入纯水1232g,再将1号反应釜中的物料加入5号还原釜中。控制温度为11℃,加入锌粉4g,搅拌5min。
再在真空条件下慢慢抽入23%的稀硫酸溶液600g,然后在5℃的温度条件下慢慢加入207g锌粉,加锌粉的时间控制为40min,锌粉加完后,搅拌反应30min,得到产物舒巴坦酸。
s4、萃取并干燥,具体如下:
反应结束后,过滤反应所得的产物舒巴坦酸,取滤液转移至6号还原釜中,静置分层。将水相转移至7号萃取釜中,采用507g的乙酸乙酯分三次萃取,每次169g,萃取完后再将7号萃取釜中的有机相以及5号还原釜中剩余的有机相合并至8号脱色釜中,加入活性炭8.5g,在室温下脱色30min,然后进行过滤,取滤液转移至9号结晶釜中。
控制釜内温度为26℃,控制压力为-0.09mpa,对9号结晶釜中的滤液进行减压蒸馏操作,蒸馏至滤液析出结晶。并注意观察滤液的状态,当蒸馏浓缩溶液逐渐开始发白时,注意观察固体物料的状态,当固体物料比较黏稠,流动性比较顺畅时,停止浓缩蒸馏。
将蒸馏所得的固体物料放入离心机中离心甩干,然后用141g乙酸乙酯泡洗再甩干。
将离心物料投入双锥干燥器中,先室温干燥1h,再打开热水系统,升温至43℃,待温度稳定后,再恒温干燥5h。
关掉热水,再抽真空0.5h,并取样送化验测水分含量,当水分含量不大于0.5%时,即可出料分装,得到成品195g,总收率71%。
比较例4
一种舒巴坦酸的合成方法,包括以下步骤:
s1、重氮化及溴化反应,具体如下:
在1号反应釜中加入乙酸乙酯852g,降温至5℃,以50r/min的转速进行搅拌,边搅拌边滴加溴素400g(2.5mol)以及纯水430g,控制温度为5℃,再滴加25%的稀硫酸980g(2.5mol)。
控制温度为8℃,以250r/min的转速进行搅拌,边搅拌边加入亚硝酸钠固体113.5g,亚硝酸钠固体分5次加入,加入时间为20min。
将216g(1mol)的6-氨基青霉烷酸用真空上料机从投料口加入1号反应釜中,控制温度为30℃,保温60min,反应结束。
再向1号反应釜中滴加亚硫酸氢钠溶液(750g水+375g亚硫酸氢钠),控制温度为15℃,使物料颜色褪色。物料为淡黄色,用淀粉-碘化钾指示剂检测,当滴加至淀粉-碘化钾指示剂5秒内不变蓝时,停止滴加。
静置分层,将水相转移至2号萃取釜中,采用540g的乙酸乙酯分三次萃取,每次用180g。萃取完成后再将2号萃取釜中的有机相转移至1号反应釜中,并向1号反应釜中滴加186g纯水进行洗涤,洗涤完成后用稀碱液(625g水+187g氢氧化钠)调节ph至7.1,并控制温度为12℃,得到第一中间体。
s2、氧化反应,具体如下:
配制氧化液:在真空条件下往3号反应釜中加入纯水1000g、25%的稀硫酸溶液705.6g(1.8mol),再通过投料口投入高锰酸钾316g(2mol),并搅拌溶解以形成氧化液。
将1号反应釜中的第一中间体冷却至15℃,在20℃的温度条件下向1号反应釜中滴加氧化液,滴加时间为1h。加完在12℃的温度条件下反应30min。
再将1号反应釜中的温度降至5℃,并向1号反应釜中滴加15%的亚硫酸氢钠溶液,直至物料褪色。
静置分层,将水层转移至4号萃取釜中,采用540g的乙酸乙酯分三次萃取,每次用180g。萃取完后再将4号萃取釜中的有机相转移至1号反应釜中,并向1号反应釜中滴加210g的纯水进行洗涤,得到第二中间体。
s3、氢化反应,具体如下:
在5号还原釜中加入纯水1300g,再将1号反应釜中的物料加入5号还原釜中。控制温度为12℃,加入锌粉4g,搅拌5min。
再在真空条件下慢慢抽入25%的稀硫酸溶液488g,然后在10℃的温度条件下慢慢加入211g锌粉,加锌粉的时间控制为50min,锌粉加完后,搅拌反应30min,得到产物舒巴坦酸。
s4、萃取并干燥,具体如下:
反应结束后,过滤反应所得的产物舒巴坦酸,取滤液转移至6号还原釜中,静置分层。将水相转移至7号萃取釜中,采用540g的乙酸乙酯分三次萃取,每次180g,萃取完后再将7号萃取釜中的有机相以及5号还原釜中剩余的有机相合并至8号脱色釜中,加入活性炭8g,在室温下脱色30min,然后进行过滤,取滤液转移至9号结晶釜中。
控制釜内温度为32℃,控制压力为-0.09mpa,对9号结晶釜中的滤液进行减压蒸馏操作,蒸馏至滤液析出结晶。并注意观察滤液的状态,当蒸馏浓缩溶液逐渐开始发白时,注意观察固体物料的状态,当固体物料比较黏稠,流动性比较顺畅时,停止浓缩蒸馏。
将蒸馏所得的固体物料放入离心机中离心甩干,然后用150g乙酸乙酯泡洗再甩干。
将离心物料投入双锥干燥器中,先室温干燥1h,再打开热水系统,升温至45℃,待温度稳定后,再恒温干燥5h。
关掉热水,再抽真空0.5h,并取样送化验测水分含量,当水分含量不大于0.5%时,即可出料分装,得到成品206g,总收率75%。
根据实施例1-4与比较例1的总收率的数据对比可得,比较例1中采用传统的制备方法制得的舒巴坦酸的反应总收率为77%,实施例1-5中采用浓度为20%-25%的稀硫酸溶液参与反应,并控制6-氨基青霉烷酸与溴素的摩尔比为1:1.5-2、6-氨基青霉烷酸与稀硫酸的摩尔比为1:1-2以及高锰酸钾与稀硫酸的摩尔比为2:1-1.5,制备所得的舒巴坦酸的收率达90%以上,由此可得,通过采用20%-25%的稀硫酸溶液以及控制各反应物的摩尔比,有利于提高反应的转化率,使得反应的收率提高,进而有利于提高反应的总收率。
根据实施例1-4与比较例2的总收率的数据对比可得,比较例2中采用30%的稀硫酸溶液进行反应,在其他条件以及操作与实施例1相同的情况下,制备所得的舒巴坦酸的总收率为78%,而实施例1-5中采用浓度为20%-25%的稀硫酸溶液进行反应,反应收率均大于90%,由此可得,通过采用20%-25%的稀硫酸溶液有利于提高反应的收率,使得反应的总收率提高。
根据实施例1-4与比较例3-4的总收率的数据对比可得,6-氨基青霉烷酸与溴素在比较例3中的摩尔比为1:1.2,在比较例4中的摩尔比为1:2.5;6-氨基青霉烷酸与稀硫酸溶液在比较例3中的摩尔比为1:0.5,在比较例4中的摩尔比为1:2.5;高锰酸钾与稀硫酸溶液的在比较例3中的摩尔比为2:0.5,在比较例4中的摩尔比为2:1.8;而采用比较例3的制备方法制备所得的舒巴坦酸的总收率为71%,采用比较例4的制备方法制备所得的舒巴坦酸的总收率为75%,采用实施例1-4的方法制备所得的舒巴坦酸的总收率均大于90%,由此可得,通过控制6-氨基青霉烷酸与溴素的摩尔比为1:1.5-2、6-氨基青霉烷酸与稀硫酸溶液的摩尔比为1:1.2以及高锰酸钾与稀硫酸溶液的摩尔比为2:1-1.5,有利于提高反应的收率,使得反应的总收率大大提高。
本具体实施方式的实施例均为本发明的较佳实施例,并非依此限制本发明的保护范围,故:凡依本发明的结构、形状、原理所做的等效变化,均应涵盖于本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!