泊沙康唑杂质及其制备方法与应用与流程
本发明属于抗感染技术领域,具体涉及一种泊沙康唑杂质及其制备方法与应用。
背景技术:
泊沙康唑2005年10月在欧洲首次上市,商品名为
泊沙康唑的化学结构式如式v所示:
本领域已知,处于对人体给药安全考虑,在一种有效的药物成分(api)产品商业化之前需要由国家和国际的管理建立毒理学上非特征性杂质的鉴定的极低下限。通常每种杂质的限量少于约0.15%重量比。未鉴定的和/或非特征性杂质的限度明显更低,通常少于0.1%重量比。
本领域中也已知,泊沙康唑任何有效成分(api)中的杂质可能来自于api本身的降解和制造过程,包括化学合成。工艺杂质包括未反应的原材料、原材料中所含的杂质及其化学衍生物、合成副产物和降解产物。
关于泊沙康唑的杂质,文献(structuralcharacterizationofimpuritiesanddegradationproducts164-177)有过报道,但对于泊沙康唑在制备过程中形成的副反应的相应工艺杂质,目前未有涉及。
技术实现要素:
本发明的目的是为了克服现有技术中存在的上述问题,提供一种泊沙康唑杂质,即式i化合物、式ii化合物。
本发明的另一个目的是提供所述式i化合物、式ii化合物的制备方法。
本发明的最后一个目的是提供所述式i化合物、式ii化合物的应用。
为实现上述目的,本发明提供的技术方案如下:
本发明所述的具有式i所示结构的化合物:
其化学名为4-(4-(4-(4-(((3r,5r)-5-((1h-1,2,4-三唑-1-基)甲基)-5-(2,4-二氟苯基)四氢呋喃-3-基)甲氧基)-3-(4-(4-(4-(1-((2s,3s)-2-(苄氧基)戊基-3-基)-5-氧-1h-1,2,4-三唑-4(5h)-基)苯基)哌嗪基-1-基)苯氧基)苯基)哌嗪基-1-基)苯基)-1-((2s,3s)-2-(苄氧基)戊基-3-基)-1h-1,2,4-三唑-5(4h)-酮。
所述式i化合物在一些实施方案中被分离为具有至少50%的纯度,或者至少60%的纯度,或者至少70%的纯度,或者至少80%的纯度,或者至少90%的纯度,或者至少95%的纯度,或者至少96%的纯度,或者至少97%的纯度,或者至少98%的纯度,或者至少99%的纯度,该纯度一般为hplc面积归一化的纯度。
本发明所述的具有式ii所示结构的化合物:
其化学名为n-(4-(4-(4-(((3s,5s)-5-((1h-1,2,4-三唑-1-基)甲基)-5-(2,4-二氟苯基)四氢呋喃-3-基)甲氧基)苯基)哌嗪基-1-基)苯基)氰胺。
式ii化合物在一些实施方案中被分离为具有至少50%的纯度,或者至少60%的纯度,或者至少70%的纯度,或者至少80%的纯度,或者至少90%的纯度,或者至少95%的纯度,或者至少96%的纯度,或者至少97%的纯度,或者至少98%的纯度,或者至少99%的纯度,该纯度一般为hplc面积归一化的纯度。
本发明所述的具有式i所示结构的化合物的制备方法,包括:将式iv化合物与式iii化合物在碱性条件下溶剂中反应得到式i化合物,
其中,式iv化合物与式iii化合物的摩尔比为(1.0-1.5):1,优选(1.1-1.3):1。
其中,碱性试剂是氢氧化钠、氢氧化钾或氢氧化锂,碱性试剂摩尔用量为式iii化合物的0.8-1.5倍,优选1.0-1.2倍。
优选地,所述碱性试剂是氢氧化钠、氢氧化钾或氢氧化锂的水溶液,所述氢氧化锂水溶液优选一水合氢氧化锂水溶液。碱性试剂摩尔用量(以碱性物质计)为式iii化合物的0.8-1.5倍,优选1.0-1.2倍。
其中,所述溶剂是n,n-二甲基甲酰胺、n-甲基吡咯烷酮、二甲亚砜或n,n-二甲基乙酰胺,反应温度为20~70℃,优选40~60℃,进一步优选45~55℃。反应时间是5~10h,优选7h。
本发明所述的具有式ii所示结构的化合物的制备方法,包括:将式iv化合物与式iii化合物在碱性条件下溶剂中反应得到式ii化合物,
其中,式iv化合物与式iii化合物的摩尔比为(1.0-1.5):1,优选(1.1-1.3):1。
其中,碱性试剂是氢氧化钠、氢氧化钾或氢氧化锂。碱性试剂摩尔用量为式iii化合物的1.0-3.0倍,优选2.0-3.0倍。
优选地,所述碱性试剂是氢氧化钠、氢氧化钾或氢氧化锂的水溶液,优选是氢氧化钠水溶液。碱性试剂摩尔用量(以碱性物质计)为式iii化合物的1.0-3.0倍,优选2.0-3.0倍。
其中,所述溶剂是n,n-二甲基甲酰胺、n-甲基吡咯烷酮、二甲亚砜或n,n-二甲基乙酰胺,反应温度为40~100℃,优选60~80℃。反应时间是3~6h,优选4h。
最终的式i化合物和式ii化合物用hplc分析其纯度,并采用核磁共振谱和质谱确证其结构。
本发明所述的式i化合物或式ii化合物在制备泊沙康唑原料药或其制剂的杂质对照品中的应用。
本发明提供了二种泊沙康唑工艺杂质及其制备方法。本发明所述的化合物i和化合物ii可以作为杂质对照品,在泊沙康唑原料和其制剂的质量研究得到应用。
有益效果:1、本发明提供了两种新化合物,作为泊沙康唑杂质对照品更好地控制原料药或制剂质量。2、本发明提供了上述两种新化合物的制备方法,该方法操作简单,反应条件温和,绿色环保,且收率高、纯度高。
附图说明
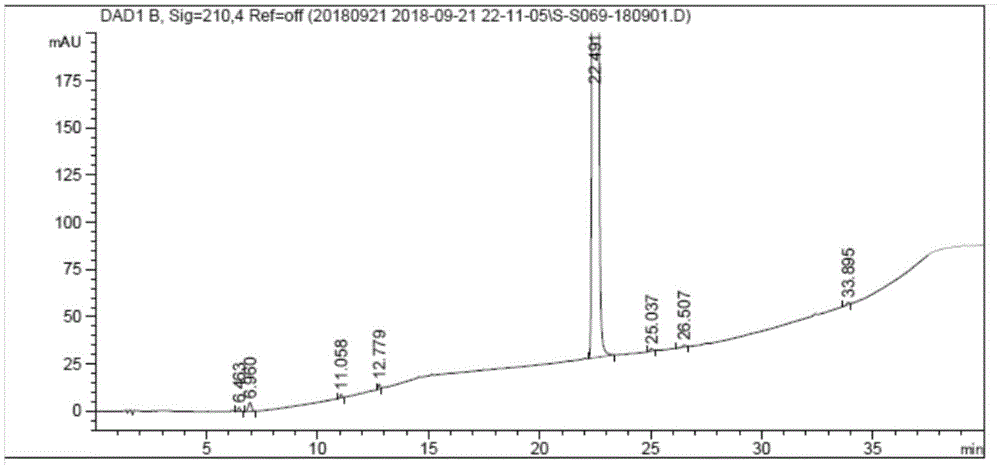
图1是中间体vi的hplc图谱。
具体实施方式
本发明将于下文通过实施例更加详细的描述,这些实施例示例性地用于进一步说明,且不应当视为对本发明的限制。
实施例1:式i化合物的制备
将式iv化合物0.0111mol(10g)与式iii化合物0.0097mol(10g)加入n,n-二甲基甲酰胺50ml中,升温至50-55℃,加入一水合氢氧化锂水溶液(1.05g一水合氢氧化锂溶于3ml纯化水中),搅拌反应7小时后,加入二氯甲烷50ml和纯化水50ml,萃取后静置分液,有机相再用纯化水水洗三次,得到的有机相浓缩后用柱层析方法进行纯化分离,得式i化合物固体。hplc纯度:95%。(ms:m/z=651.9[m+2h]2+;1h-nmr(400mhz,dmso-d6)δ:8.32(s,1h),8.31(s,1h),8.25(s,1h),7.73(s,1h),7.47(dd,4h),7.22(m,7h),7.16(m,5h),7.11(dd,4h),6.95(m,4h),6.78(m,3h),6.72(d,1h),4.52(d,2h),4.45(t,2h),4.25(d,2h),3.98(dd,2h),3.80(m,2h),3.70(m,3h),3.53(t,1h),3.31(m,8h),3.19(m,8h),2.35(m,1h),2.22(m,1h),1.93(m,1h),1.74(m,4h),1.23(d,6h),0.78(t,6h);13c-nmr(400mhz,dmso-d6)δ:,152.5,151.4,150.4,149.7,146.3,145.5,144.9,143.4,138.6,134.5,127.9,127.1,125.4,122.8,117.3,116.8,115.6,112.0,110.0,83.3,75.7,69.8,60.6,55.2,49.1,48.9,48.1,38.3,37.6,21.5,16.3,10.4。)
实施例2:式ii化合物的制备
将式iv化合物0.0111mol(5.0g)与式iii化合物0.0097mol(5.0g)加入二甲亚砜100ml中,加入氢氧化钠水溶液(0.97g氢氧化钠溶于2ml纯化水中),升温至75-80℃,搅拌反应4小时后,加入乙酸乙酯100ml和纯化水50ml,萃取后静置分液除去有机相,水相转移至反应瓶中搅拌析晶48h后过滤收集固体得式ii化合物。hplc纯度:96%。(ms:m/z=572.3[m+h]+;1h-nmr(400mhz,dmso-d6)δ:8.35(s,1h),7.79(s,1h),7.30(m,2h),7.00(m,3h),6.92(m,2h),6.86(m,2h),6.79(m,2h),4.57(dd,2h),4.00(t,1h),3.74(m,2h),3.66(t,1h),3.16(m,4h),3.14(m,4h),2.52(m,1h),2.39(m,1h),2.12(m,1h);13c-nmr(400mhz,dmso-d6)δ:163.0,160.6,160.0,157.5,152.1,150.5,146.8,145.4,145.0,130.7,128.4,126.2,117.5,117.4,115.8,115.0,112.9,111.0,104.5,83.3,69.9,68.7,55.2,49.6,49.2,38.4,37.5。
实施例3:式i化合物的制备
与实施例1相同,区别仅在于:
式iv化合物与式iii化合物的摩尔比为1:1。碱性试剂是氢氧化钠水溶液,碱性试剂摩尔用量(以碱性物质计)为式iii化合物的0.8倍。溶剂是n-甲基吡咯烷酮,反应温度为20℃。制备得到式i化合物。
实施例4:式i化合物的制备
与实施例1相同,区别仅在于:
式iv化合物与式iii化合物的摩尔比为1.5:1。碱性试剂是氢氧化钾水溶液,碱性试剂摩尔用量(以碱性物质计)为式iii化合物的1.5倍。溶剂是二甲亚砜,反应温度为70℃。制备得到式i化合物。
实施例5:式ii化合物的制备
与实施例2相同,区别仅在于:
式iv化合物与式iii化合物的摩尔比为1.0:1。碱性试剂是氢氧化钾水溶液,碱性试剂摩尔用量(以碱性物质计)为式iii化合物的1.0倍。溶剂是n,n-二甲基甲酰胺,反应温度为40~100℃。制备得到式ii化合物。
实施例6:式ii化合物的制备
与实施例2相同,区别仅在于:
式iv化合物与式iii化合物的摩尔比为1.5:1。碱性试剂是氢氧化锂水溶液,碱性试剂摩尔用量(以碱性物质计)为式iii化合物的3.0倍。溶剂是n-甲基吡咯烷酮,反应温度为40~100℃。制备得到式ii化合物。
实施例7:
参照专利cn201110185639.8报道的合成方法(详见合成路线1)制备泊沙康唑。
合成路线1:
申请人在泊沙康唑制备研究过程中,发现了在采用式iv化合物与式iii化合物合成泊沙康唑关键中间体vi过程中,多批次样品中均检出杂质式i化合物和式ii化合物(见图1,其中式i化合物为rt=33.895min,式ii化合物为rt=6.960min,中间体vi为rt=22.491min),使得最终产品质量难以保证。
图1的检测色谱条件为:十八硅烷键合硅胶为填充剂;流动相a为10mmol/l磷酸氢二钾(0.1%磷酸调节ph至9.2),流动相b为甲醇:乙腈=1:1;流速为每分钟1.0ml,线性梯度洗脱。
- 还没有人留言评论。精彩留言会获得点赞!