一种分离提纯甘木通有效成分的方法与流程
本发明涉及化学提纯领域,具体涉及一种分离提纯甘木通有效成分的方法。
背景技术:
甘木通(clematisfilamentosadunn),别名眼蛇药,毛茛科铁线莲属,两广分布,以叶茎入药,味甘微凉。民间用于治疗红眼头痛等,有降压,镇静,催眠,扩张血管及改善血循环的效果。被认为是一种无毒性,安全性高,及药源丰富的治疗冠心病和高血压的植物药。以其为原料的单方中成药冠心康片临床用于冠心病、高血压等的治疗。
有文献报道称,在对甘木通进行提取后发现,其提取物中的主要活性成分为黄酮类化合物。黄酮类化合物是一类具有重要药用价值的化学骨架,此类骨架具有多种活性功能,尤其对心肌缺血具有明显的治疗作用。目前对甘木通中成分的分离提纯通常先将甘木通的地上部分粉碎后,使用乙醇浸泡提纯。然而这种提纯方式还停留在粗提取的程度,粗提取物为多种物质的混合物,其中还含有多种非活性杂质,利用常规的分离方法进行进一步分离时发现,tlc(薄层色谱)上的点重叠在一起,液相色谱也出现重叠峰,难以将活性物质逐一单独地分离出来,使得其所含的各种黄酮类化合物的具体构成和结构尚不明确,无法得知其在心肌缺血治疗上的确切作用机制。
因此,对甘木通进行精提纯,得到其纯净的活性物质,对研究分析其治疗心肌缺血的机理有着重要的意义,而且提纯后得到的活性物质可进一步用于制作成药物,为冠心病和高血压患者带来治疗。
技术实现要素:
本发明的目的在于提供一种分离提纯甘木通有效成分的方法,该方法可以将甘木通中的活性物质逐一单独提纯出来,且提纯后的物质纯度高。
本发明的目的通过以下技术方案实现:
一种分离提纯甘木通有效成分的方法,包括以下步骤:
(1)使用乙醇对甘木通的地上部分进行提取,获得乙醇提取物;
(2)将所述乙醇提取物上正相硅胶色谱柱,依次使用体积比为11:1~9:1、8:1~6:1、4:1~2:1、1.5:1~0.5:1的石油醚/丙酮混合溶剂、丙酮以及甲醇进行梯度洗脱,并分别收集洗脱液并除去溶剂,得到提取物a1、a2、a3、a4、a5、a6;
(3)将提取物a2上正相硅胶柱色谱,依次使用体积比为100:1~80:1和60:1~40:1的氯仿/甲醇混合溶剂进行梯度洗脱,收集60:1~40:1的氯仿/甲醇的洗脱液并蒸发溶剂浓缩后,上葡聚糖凝胶色谱柱sephadexlh-20,用体积比为2:1~0.5:1的氯仿/甲醇混合溶剂洗脱,静置洗脱液析出粉末状沉淀,过滤分离,干燥后得到鼠尾草素(cas号:19103-54-9);
(4)将提取物a3上正相硅胶色谱柱,使用体积比为40:1~20:1的氯仿/甲醇混合溶剂进行洗脱,先后分两批次收集得到洗脱液t1和t2,蒸发溶剂浓缩后得到提取物f1和提取物f2;
(4a)将提取物f1上葡聚糖凝胶色谱柱sephadexlh-20,用体积比为2:1~0.5:1的氯仿/甲醇混合溶剂进行洗脱,静置洗脱液析出粉末状沉淀,过滤分离得到齐墩果酸(cas号:508-02-1);
(4b)将提取物f2上葡聚糖凝胶色谱柱sephadexlh-20,用体积比为2:1~0.5:1的氯仿/甲醇混合溶剂进行洗脱,通过hplc制备色谱法进行处理,分别得到提纯液t3和t4,除去溶剂并干燥后分别得到落叶松树脂醇(cas号:27003-73-2)和二氢脱氢二松柏醇(dihydrodehydrodiconiferylalcohol,cas号:126253-41-6);
(5)将提取物a5上正相硅胶色谱柱,用体积比为25:1~5:1的氯仿/甲醇混合溶剂进行洗脱,先后分两批次收集得到洗脱液t5和t6,浓缩后得到提取物f3和f4;
(5a)将提取物f3上葡聚糖凝胶色谱柱sephadexlh-20,用体积比为2:1~0.5:1的氯仿/甲醇混合溶剂进行洗脱,静置洗脱液析出粉末状沉淀,固液分离,用有机溶剂将粉末状沉淀溶解后,上mcigelchp20/p120柱,用60~80%甲醇洗脱,收集洗脱液,浓缩,重结晶并干燥后得到芹菜素(cas号:520-36-5);
(5b)将提取物f4上mcigelchp20/p120柱,用70~90%甲醇洗脱,收集洗脱液,浓缩,重结晶并干燥后得到木犀草素(cas号:491-70-3);
(6)将提取物a6浓缩后,加水溶解,然后加入与加水后液体总体积等体积的正丁醇进行多次萃取,合并正丁醇相并浓缩后上反相硅胶色谱柱,依次使用10~20%、20~35%和35~50%的甲醇水溶液进行洗脱,收集洗脱液,浓缩并干燥后得到提取物f5、f6和f7;
(6a)将提取物f5进行浓缩后上葡聚糖凝胶色谱柱sephadexlh-20,用体积比为2:1~0.5:1的氯仿/甲醇混合溶剂进行洗脱,收集洗脱液并浓缩后上正相硅胶色谱柱,使用体积比为9:1~7:1的氯仿/甲醇混合溶剂进行洗脱,收集洗脱液,浓缩并干燥后,得到咖啡酸(cas号:331-39-5);
(6b)将提取物f6进行浓缩后上葡聚糖凝胶色谱柱sephadexlh-20,用体积比为2:1~0.5:1的氯仿/甲醇混合溶剂进行洗脱,收集洗脱液并浓缩后上正相硅胶色谱柱,使用体积比为7:1~5:1的氯仿/甲醇混合溶剂进行洗脱,收集洗脱液,浓缩并干燥后,得到urolignoside(cas号:131723-83-6);
(6c)将提取物f7进行浓缩后上葡聚糖凝胶色谱柱sephadexlh-20,用体积比为2:1~0.5:1的氯仿/甲醇混合溶剂进行洗脱,先后分两批次收集洗脱液t7和t8,浓缩干燥后分别得到紫云英苷(cas号:480-10-4)和蒙花苷(cas号:480-36-4)。
本发明所述的甘木通的地上部分包括甘木通的茎枝。
为了克服常规的分离方法无法逐一分离甘木通中有效成分的问题,本发明经过长期实验后,以不同种类的色谱柱结合hplc和萃取等分离方法,经过分步提纯,在物质组成复杂的甘木通乙醇粗提取物中,将其中具有价值的有效成分鼠尾草素、齐墩果酸、落叶松树脂醇、二氢脱氢二松柏醇、芹菜素、木犀草素、咖啡酸、urolignoside、紫云英苷和蒙花苷的高效地实现分离提纯,确定了甘木通中黄酮类化合物的具体组成,提纯后的物质纯度可达95%以上,在科研和医药领域都具有重要的意义。
本发明步骤(2)的洗脱过程中,用11:1~9:1的石油醚/丙酮混合溶剂洗脱得到的为提取物a1;用8:1~6:1的石油醚/丙酮混合溶剂洗脱得到的为提取物a2;用4:1~2:1的石油醚/丙酮混合溶剂洗脱得到的为提取物a3;用1.5:1~0.5:1的石油醚/丙酮混合溶剂洗脱得到的为提取物a4;用丙酮洗脱得到的为提取物a5;用甲醇提纯洗脱得到的为提取物a6,其中提取物a1和a4为杂质成分。在步骤(4)、(5)和(6c)中所述的先后分两批次收集洗脱液,应理解为,使用tlc进行验证,将先洗脱物的洗脱液收集并合并成为第一批次,再将后洗脱物的洗脱液收集合并成为第二批次,以收集得到两种不同的提取物。
作为本发明的一种实施方式,步骤(1)对甘木通的地上部分进行提取的过程具体为:将干燥后的甘木通地上部分进行粉碎,按固体质量/液体体积比1:3~1:5的比例加入85%工业乙醇,提取三次,合并上清液,真空浓缩并-80℃条件下进行冷藏。
优选地,步骤(2)依次使用体积比为10:1、7:1、3:1、1:1的石油醚/丙酮混合溶剂、丙酮以及甲醇进行梯度洗脱。
优选地,步骤(3)将提取物a2上正相硅胶柱色谱,依次使用体积比为100:1和50:1的氯仿/甲醇混合溶剂进行梯度洗脱,收集50:1的氯仿/甲醇的洗脱液并蒸发溶剂浓缩后,上葡聚糖凝胶色谱柱sephadexlh-20,用体积比为1:1的氯仿/甲醇混合溶剂洗脱。
优选地,步骤(4)使用体积比为30:1的氯仿/甲醇混合溶剂进行洗脱。
优选地,步骤(4a)用体积比为1:1的氯仿/甲醇混合溶剂进行洗脱。
优选地,步骤(4b)用体积比为1:1的氯仿/甲醇混合溶剂进行洗脱。
优选地,步骤(5)用体积比为15:1的氯仿/甲醇混合溶剂进行洗脱。
优选地,步骤(5a)将提取物f3上葡聚糖凝胶色谱柱sephadexlh-20,用体积比为1:1的氯仿/甲醇混合溶剂进行洗脱,静置洗脱液析出粉末状沉淀,固液分离,用有机溶剂将粉末状沉淀溶解后,上mcigelchp20/p120柱,用75%甲醇洗脱。
优选地,步骤(5b)用80%甲醇洗脱。
优选地,步骤(6a)将提取物f5进行浓缩后上葡聚糖凝胶色谱柱sephadexlh-20,用体积比为1:1的氯仿/甲醇混合溶剂进行洗脱,收集洗脱液并浓缩后上正相硅胶色谱柱,使用体积比为8:1的氯仿/甲醇混合溶剂进行洗脱。
优选地,步骤(6b)将提取物f6进行浓缩后上葡聚糖凝胶色谱柱sephadexlh-20,用体积比为1:1的氯仿/甲醇混合溶剂进行洗脱,收集洗脱液并浓缩后上正相硅胶色谱柱,使用体积比为6:1的氯仿/甲醇混合溶剂进行洗脱。
优选地,步骤(6c)用体积比为1:1的氯仿/甲醇混合溶剂进行洗脱。
本发明推荐在所述步骤(2)中使用直径3.5cm,长40cm玻璃柱的硅胶色谱柱;步骤(3)、(4)、(5)中使用直径3.5cm,长40cm玻璃柱的硅胶色谱柱。
作为本发明的一种实施方式:
所述步骤(3)中将粉末状沉淀过滤分离后,使用甲醇进行洗涤;
所述步骤(4a)中将粉末状沉淀过滤分离后,使用丙酮进行洗涤;
所述步骤(4b)使用hplc制备色谱法的过程中,溶剂为30~50%的甲醇水溶液;
所述步骤(5a)所述用于溶解粉末状沉淀的有机溶剂为dmso;
所述步骤(5a)、(5b)使用甲醇进行重结晶;
本发明所述的干燥采用冷冻干燥。
本发明与现有技术相比具有以下有益效果:
1.本发明在分离提纯的过程中,使用正相硅胶柱进行梯度粗分离后,采用葡聚糖凝胶色谱或结合高效液相色谱分离的方式,实现了鼠尾草素、齐墩果酸、落叶松树脂醇和二氢脱氢二松柏醇的快速高效分离,纯度可达95%以上;
2.本发明采用葡聚糖凝胶色谱与mci色谱相结合或mci色谱柱单一分离的方式,实现了黄酮类化合物芹菜素和木犀草素快速高效的分离,纯度均可达到95%以上,克服了高效液相色谱成分峰分离困难的技术难题;
3.本发明使用葡聚糖凝胶色谱或结合正相硅胶柱恒定洗脱后,实现了咖啡酸、urolignoside、紫云英苷和蒙花苷的快速高效分离,纯度可达95%以上,克服了高效液相色谱成分峰分离困难的技术难题;
4.本发明对甘木通中的活性物质实现有效的逐一分离,提纯得到纯净的化合物,有助于科学研究,以确定其治疗心血管作用物质基础、鉴定其单体的化学结构、明析其作用机理,也可以将提纯工艺可放大,将提纯后的活性成分用于制成降低慢性疾病发生发展的药物,应用领域广阔,市场潜力巨大,实现甘木通的二次开发实现升级和质量控制,这对促进甘木通资源的发展、提高甘木通产业的经济效益、推动甘木通产业的可持续发展具有重要意义。
附图说明
以下通过附图对本发明作进一步的说明。
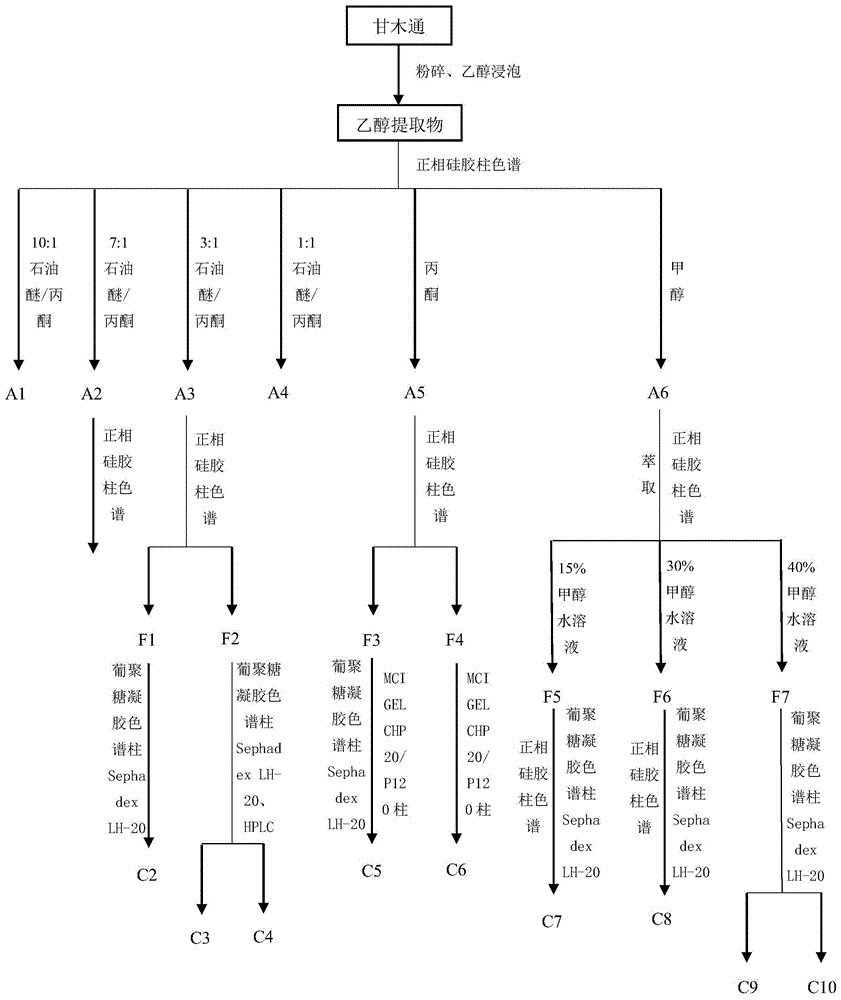
图1实施例3分离提纯流程示意图。
图2化合物c1的nmr氢谱图。
图3化合物c1的nmr碳谱图。
图4化合物c1的分子结构式。
图5化合物c2的nmr氢谱图。
图6化合物c2的nmr碳谱图。
图7化合物c2的分子结构式。
图8化合物c3的nmr氢谱图。
图9化合物c3的nmr碳谱图。
图10化合物c3的分子结构式。
图11化合物c4的nmr氢谱图。
图12化合物c4的nmr碳谱图。
图13化合物c4的分子结构式。
图14化合物c5的nmr氢谱图。
图15化合物c5的nmr碳谱图。
图16化合物c5的分子结构式。
图17化合物c6的nmr氢谱图。
图18化合物c6的nmr碳谱图。
图19化合物c6的分子结构式。
图20化合物c7的nmr氢谱图。
图21化合物c7的分子结构式。
图22化合物c8的nmr氢谱图。
图23化合物c8的nmr碳谱图。
图24化合物c8的分子结构式。
图25化合物c9的nmr氢谱图。
图26化合物c9的nmr碳谱图。
图27化合物c9的分子结构式。
图28化合物c10的nmr氢谱图。
图29化合物c10的分子结构式。
具体实施方式
以下通过具体的实施例对本发明作进一步的说明。
实施例1
(1)将10kg干燥后的甘木通地上部分进行粉碎,按固体质量/液体体积比1:5的比例加入85%工业乙醇,浸泡提取12h,提取三次,合并上清液,真空浓缩并-80℃条件下进行冷藏,获得乙醇提取物;
(2)将所述乙醇提取物上正相硅胶色谱柱,依次使用体积比为11:1、8:1、4:1、1.5:1的石油醚/丙酮混合溶剂、丙酮以及甲醇进行梯度洗脱,并分别收集洗脱液并除去溶剂,得到提取物a1、a2、a3、a4、a5、a6;
(3)将提取物a2上正相硅胶柱色谱,依次使用体积比为90:1和60:1的氯仿/甲醇混合溶剂进行梯度洗脱,收集60:1的氯仿/甲醇的洗脱液并蒸发溶剂浓缩后,上葡聚糖凝胶色谱柱sephadexlh-20,用体积比为2:1的氯仿/甲醇混合溶剂洗脱,静置洗脱液析出粉末状沉淀,过滤分离,使用甲醇进行洗涤,干燥后得到化合物c1;
(4)将提取物a3上正相硅胶色谱柱,使用体积比为40:1的氯仿/甲醇混合溶剂进行洗脱,先后分两批次收集得到洗脱液t1和t2,蒸发溶剂浓缩后得到提取物f1和提取物f2;
(4a)将提取物f1上葡聚糖凝胶色谱柱sephadexlh-20,用体积比为2:1的氯仿/甲醇混合溶剂进行洗脱,静置洗脱液析出粉末状沉淀,使用丙酮进行洗涤,过滤分离得到化合物c2;
(4b)将提取物f2上葡聚糖凝胶色谱柱sephadexlh-20,用体积比为2:1的氯仿/甲醇混合溶剂进行洗脱,通过hplc制备色谱法进行处理,溶剂为30%的甲醇水溶液;,分别得到提纯液t3和t4,除去溶剂并干燥后分别得到化合物c3和化合物c4;
(5)将提取物a5上正相硅胶色谱柱,用体积比为25:1的氯仿/甲醇混合溶剂进行洗脱,先后分两批次收集得到洗脱液t5和t6,浓缩后得到提取物f3和f4;
(5a)将提取物f3上葡聚糖凝胶色谱柱sephadexlh-20,用体积比为2:1的氯仿/甲醇混合溶剂进行洗脱,静置洗脱液析出粉末状沉淀,固液分离,用dmso将粉末状沉淀溶解后,上mcigelchp20/p120柱,用60%甲醇洗脱,收集洗脱液,浓缩,在甲醇中重结晶并干燥后得到c5;
(5b)将提取物f4上mcigelchp20/p120柱,用70%甲醇洗脱,收集洗脱液,浓缩,在甲醇中重结晶并干燥后得到c6;
(6)将提取物a6浓缩后,加水溶解,然后加入与加水后液体总体积等体积的正丁醇进行多次萃取,合并正丁醇相并浓缩后上反相硅胶色谱柱,依次使用10%、20%和35%的甲醇水溶液进行洗脱,收集洗脱液,浓缩并干燥后得到提取物f5、f6和f7;
(6a)将提取物f5进行浓缩后上葡聚糖凝胶色谱柱sephadexlh-20,用体积比为2:1的氯仿/甲醇混合溶剂进行洗脱,收集洗脱液并浓缩后上正相硅胶色谱柱,使用体积比为9:1的氯仿/甲醇混合溶剂进行洗脱,收集洗脱液,浓缩并干燥后,得到化合物c7;
(6b)将提取物f6进行浓缩后上葡聚糖凝胶色谱柱sephadexlh-20,用体积比为2:1的氯仿/甲醇混合溶剂进行洗脱,收集洗脱液并浓缩后上正相硅胶色谱柱,使用体积比为7:1的氯仿/甲醇混合溶剂进行洗脱,收集洗脱液,浓缩并干燥后,得到化合物c8;
(6c)将提取物f7进行浓缩后上葡聚糖凝胶色谱柱sephadexlh-20,用体积比为2:1的氯仿/甲醇混合溶剂进行洗脱,先后分两批次收集洗脱液t7和t8,浓缩干燥后分别得到化合物c9和化合物c10。
实施例2
(1)将15kg干燥后的甘木通地上部分进行粉碎,按固体质量/液体体积比1:4的比例加入85%工业乙醇,浸泡提取12h,提取三次,合并上清液,真空浓缩并-80℃条件下进行冷藏,获得乙醇提取物;
(2)将所述乙醇提取物上正相硅胶色谱柱,依次使用体积比为9:1、6:1、2:1、0.5:1的石油醚/丙酮混合溶剂、丙酮以及甲醇进行梯度洗脱,并分别收集洗脱液并除去溶剂,得到提取物a1、a2、a3、a4、a5、a6;
(3)将提取物a2上正相硅胶柱色谱,依次使用体积比为80:1和40:1的氯仿/甲醇混合溶剂进行梯度洗脱,收集40:1的氯仿/甲醇的洗脱液并蒸发溶剂浓缩后,上葡聚糖凝胶色谱柱sephadexlh-20,用体积比为0.5:1的氯仿/甲醇混合溶剂洗脱,静置洗脱液析出粉末状沉淀,过滤分离,使用甲醇进行洗涤,干燥后得到化合物c1;
(4)将提取物a3上正相硅胶色谱柱,使用体积比为20:1的氯仿/甲醇混合溶剂进行洗脱,先后分两批次收集得到洗脱液t1和t2,蒸发溶剂浓缩后得到提取物f1和提取物f2;
(4a)将提取物f1上葡聚糖凝胶色谱柱sephadexlh-20,用体积比为0.5:1的氯仿/甲醇混合溶剂进行洗脱,静置洗脱液析出粉末状沉淀,使用丙酮进行洗涤,过滤分离得到化合物c2;
(4b)将提取物f2上葡聚糖凝胶色谱柱sephadexlh-20,用体积比为0.5:1的氯仿/甲醇混合溶剂进行洗脱,通过hplc制备色谱法进行处理,溶剂为50%的甲醇水溶液;,分别得到提纯液t3和t4,除去溶剂并干燥后分别得到c3和c4;
(5)将提取物a5上正相硅胶色谱柱,用体积比为5:1的氯仿/甲醇混合溶剂进行洗脱,先后分两批次收集得到洗脱液t5和t6,浓缩后得到提取物f3和f4;
(5a)将提取物f3上葡聚糖凝胶色谱柱sephadexlh-20,用体积比为0.5:1的氯仿/甲醇混合溶剂进行洗脱,静置洗脱液析出粉末状沉淀,固液分离,用dmso将粉末状沉淀溶解后,上mcigelchp20/p120柱,用80%甲醇洗脱,收集洗脱液,浓缩,在甲醇中重结晶并干燥后得到化合物c5;
(5b)将提取物f4上mcigelchp20/p120柱,用90%甲醇洗脱,收集洗脱液,浓缩,在甲醇中重结晶并干燥后得到化合物c6;
(6)将提取物a6浓缩后,加水溶解,然后加入与加水后液体总体积等体积的正丁醇进行多次萃取,合并正丁醇相并浓缩后上反相硅胶色谱柱,依次使用20%、35%和50%的甲醇水溶液进行洗脱,收集洗脱液,浓缩并干燥后得到提取物f5、f6和f7;
(6a)将提取物f5进行浓缩后上葡聚糖凝胶色谱柱sephadexlh-20,用体积比为0.5:1的氯仿/甲醇混合溶剂进行洗脱,收集洗脱液并浓缩后上正相硅胶色谱柱,使用体积比为7:1的氯仿/甲醇混合溶剂进行洗脱,收集洗脱液,浓缩并干燥后,得到化合物c7;
(6b)将提取物f6进行浓缩后上葡聚糖凝胶色谱柱sephadexlh-20,用体积比为0.5:1的氯仿/甲醇混合溶剂进行洗脱,收集洗脱液并浓缩后上正相硅胶色谱柱,使用体积比为5:1的氯仿/甲醇混合溶剂进行洗脱,收集洗脱液,浓缩并干燥后,得到化合物c8;
(6c)将提取物f7进行浓缩后上葡聚糖凝胶色谱柱sephadexlh-20,用体积比为0.5:1的氯仿/甲醇混合溶剂进行洗脱,先后分两批次收集洗脱液t7和t8,浓缩干燥后分别得到化合物c9和化合物c10。
实施例3
(1)如图1所示,将5kg干燥后的甘木通地上部分进行粉碎,按固体质量/液体体积比1:3的比例加入85%工业乙醇,浸泡提取12h,提取三次,合并上清液,真空浓缩并-80℃条件下进行冷藏,获得乙醇提取物;
(2)将所述乙醇提取物上正相硅胶色谱柱,依次使用体积比为10:1、7:1、3:1、1:1的石油醚/丙酮混合溶剂、丙酮以及甲醇进行梯度洗脱,并分别收集洗脱液并除去溶剂,得到提取物a1、a2、a3、a4、a5、a6;
(3)将提取物a2上正相硅胶柱色谱,依次使用体积比为100:1和50:1的氯仿/甲醇混合溶剂进行梯度洗脱,收集50:1的氯仿/甲醇的洗脱液并蒸发溶剂浓缩后,上葡聚糖凝胶色谱柱sephadexlh-20,用体积比为1:1的氯仿/甲醇混合溶剂洗脱,静置洗脱液析出粉末状沉淀,过滤分离,使用甲醇进行洗涤,干燥后得到化合物c1;
(4)将提取物a3上正相硅胶色谱柱,使用体积比为30:1的氯仿/甲醇混合溶剂进行洗脱,先后分两批次收集得到洗脱液t1和t2,蒸发溶剂浓缩后得到提取物f1和提取物f2;
(4a)将提取物f1上葡聚糖凝胶色谱柱sephadexlh-20,用体积比为1:1的氯仿/甲醇混合溶剂进行洗脱,静置洗脱液析出粉末状沉淀,使用丙酮进行洗涤,过滤分离得到化合物c2;
(4b)将提取物f2上葡聚糖凝胶色谱柱sephadexlh-20,用体积比为1:1的氯仿/甲醇混合溶剂进行洗脱,通过hplc制备色谱法进行处理,溶剂为40%的甲醇水溶液;,分别得到提纯液t3和t4,除去溶剂并干燥后分别得到化合物c3和化合物c4;
(5)将提取物a5上正相硅胶色谱柱,用体积比为15:1的氯仿/甲醇混合溶剂进行洗脱,先后分两批次收集得到洗脱液t5和t6,浓缩后得到提取物f3和f4;
(5a)将提取物f3上葡聚糖凝胶色谱柱sephadexlh-20,用体积比为1:1的氯仿/甲醇混合溶剂进行洗脱,静置洗脱液析出粉末状沉淀,固液分离,用dmso将粉末状沉淀溶解后,上mcigelchp20/p120柱,用75%甲醇洗脱,收集洗脱液,浓缩,在甲醇中重结晶并干燥后得到化合物c5;
(5b)将提取物f4上mcigelchp20/p120柱,用80%甲醇洗脱,收集洗脱液,浓缩,在甲醇中重结晶并干燥后得到化合物c6;
(6)将提取物a6浓缩后,加水溶解,然后加入与加水后液体总体积等体积的正丁醇进行多次萃取,合并正丁醇相并浓缩后上反相硅胶色谱柱,依次使用15%、30%和40%的甲醇水溶液进行洗脱,收集洗脱液,浓缩并干燥后得到提取物f5、f6和f7;
(6a)将提取物f5进行浓缩后上葡聚糖凝胶色谱柱sephadexlh-20,用体积比为1:1的氯仿/甲醇混合溶剂进行洗脱,收集洗脱液并浓缩后上正相硅胶色谱柱,使用体积比为8:1的氯仿/甲醇混合溶剂进行洗脱,收集洗脱液,浓缩并干燥后,得到化合物c7;
(6b)将提取物f6进行浓缩后上葡聚糖凝胶色谱柱sephadexlh-20,用体积比为1:1的氯仿/甲醇混合溶剂进行洗脱,收集洗脱液并浓缩后上正相硅胶色谱柱,使用体积比为6:1的氯仿/甲醇混合溶剂进行洗脱,收集洗脱液,浓缩并干燥后,得到化合物c8;
(6c)将提取物f7进行浓缩后上葡聚糖凝胶色谱柱sephadexlh-20,用体积比为1:1的氯仿/甲醇混合溶剂进行洗脱,先后分两批次收集洗脱液t7和t8,浓缩干燥后分别得到化合物c9和化合物c10。
提纯结束后,以dmso为溶剂,使用频率为500mhz的nmr对提纯所得的化合物c1~c10进行h图谱和c图谱的检测分析,检测结果如下:
1)如图2所示,为化合物c1的nmr氢谱图,其氢谱数据具体为:8.07(2h,d,j=9.0hz,h-2′,6′),7.12(2h,d,j=9.0hz,h-3′,5′),6.94(1h,s,h-8),6.96(1h,s,h-3),12.87(1h,s,oh-5),3.12(1h,s,och3),3.18(1h,s,och3),3.13(1h,s,och3);
如图3所示,为化合物c1的nmr碳谱图,其碳谱数据具体为:163.6(c-2),103.3(c-3),182.2(c-4),152.5(c-5),131.9(c-6),158.1(c-7),91.6(c-8),152.8(c-5),105.2(c-10),122.7(c-1’),128(c-2’,6’),114.6(c-3’,5’),162.4(c-4’),60.2(-och3),56.4(-och3),55.6(-och3)。
在对上述数据进行综合分析后,得出化合物c1的分子结构如图4所示,证实化合物c1为鼠尾草素(cas号:19103-54-9)。
2)如图5所示,为化合物c2的nmr氢谱图,其氢谱数据具体为:12.04(s,1h,cooh),5.16(t,j=3.7,3.3hz,1h,h-12),2.99(dd,j=10.6,5.1hz,2h,h-3),2.75(dd,j=13.9,4.0hz,1h,h-18),1.91(td,j=13.6,4.0hz,1h,h-16),1.81(dd,j=8.8,3.3hz,2h,h-11),1.58-1.67(m,3h,h-2,h-15,h-19),1.38-1.52(m,8h,h-1,h-2,h-6,h-7,h-9,h-16,h-21),1.07(d,j=5.9hz,3h,h-6),1.28-1.34(m,2h,h-19,h-21),1.23(td,j=9.9,3.3hz,1h,h-5),1.13(m,1h,h-19),1.03-1.07(m,1h,h-21),0.99(td,j=11.0,3.3hz,1h,h-15),1.09,0.89,0.87,0.87,0.85,0.72,0.67(s,3h);
如图6所示,为化合物c2的nmr碳谱图,其碳谱数据具体为:178.6(c-28),143.8(c-13),121.5(c-12),76.8(c-3),54.8(c-5),47.1(c-9),45.7(c-19),45.4(c-17),41.3(c-18),40.8(c-14),38.9(c-8),38.4(c-1),36.6(c-10),33.3(c-21),32.8(c-29),32.4(c-7),32.1(c-22),30.4(c-20),28.2(c-23),27.2(c-15),26.9(c-27),25.6(c-2),23.4(c-30),22.9(c-11),22.6(c-16),18.0(c-6),16.8(c-26),16.0(c-24),15.1(c-25)。
在对上述数据进行综合分析后,得出化合物c2的分子结构如图7所示,证实化合物c2为齐墩果酸(cas号:508-02-1)。
3)如图8所示,为化合物c3的nmr氢谱图,其氢谱数据具体为:6.81(4h,m,h-5,
2′,5′,6′),6.63(2h,m,h-2,6),4.72(1h,d,j=7.2hz,h-7′),4.00(1h,dd,j=8.0,7.2hz,h-9α),3.89(1h,m,h-9′α),3.70(2h,m,h-9β,9′β),2.92(1h,dd,j=12.6,4.8hz,h-7α),2.69(1h,m,h-8),2.48(1h,dd,j=12.6,10.6hz,h-7β),2.39(1h,m,h-8′),3.88(3h,s,3′-och3),3.82(3h,s,3-och3);
如图9所示,为化合物c3的nmr碳谱图,其碳谱数据具体为:147.3(c-3′),146.9(c-3),144.9(c-4′),144.0(c-4),135.3(c-1′),132.6(c-1),121.7(c-6),119.2(c-6′),114.7(c-5),114.6(c-5′),111.8(c-2),108.9(c-2′),83.0(c-7′),73.1(c-9),60.9(c-9′),56.1(3,3′-och3),52.9(c-8′),42.9(c-8),33.8(c-7)。
在对上述数据进行综合分析后,得出化合物c3的分子结构如图10所示,证实化合物c3为落叶松树脂醇(cas号:27003-73-2)。
4)如图11所示,为化合物c4的nmr氢谱图,其氢谱数据具体为(c5d5n):7.33(1h,d,j=1.8hz,h-2),7.20(1h,d,j=1.8hz,h-5),7.25(1h,dd,j=1.8,8.1hz,h-6),6.06(1h,d,j=6.8hz,h-7),3.97(1h,m,h-8),4.21(1h,m,h-9a),4.27(1h,m,h-9b),6.92(1h,brs,h-2′),7.06(1h,brs,h-6′),2.87(2h,t,j=7.6hz,h-7′),2.09(2h,m,h-8′),3.92(2h,t,j=6.4hz,h-9′),3.63(3h,s,3-och3),3.84(3h,s,3′-och3);
如图12所示,为化合物c4的nmr碳谱图,其碳谱数据具体为(c5d5n):134.4(c-1),111.4(c-2),148.6(c-3),147.9(c-4),117.0(c-5),120.3(c-6),88.9(c-7),55.6(c-8),64.9(c-9),130.7(c-1′),114.2(c-2′),145.2(c-3′),147.9(c-4′),136.7(c-5′),118.1(c-6′),33.2(c-7′),36.5(c-8′),62.0(c-9′),56.3(3-och3),56.8(3′-och3)。
在对上述数据进行综合分析后,得出化合物c4的分子结构如图13所示,证实化合物c4为二氢脱氢二松柏醇(dihydrodehydrodiconiferylalcohol,cas号:126253-41-6)。
5)如图14所示,为化合物c5的nmr氢谱图,其氢谱数据具体为:6.78(1h,s),6.49(1h,d,j=1.8hz),6.19(1h,d,j=1.8hz),6.94(2h,dd,j=9.0,1.2hz),7.94(2h,dd,j=9.0,1.2hz);
如图15所示,为化合物c5的nmr碳谱图,其碳谱数据具体为:182.0(c-4),164.2(c-2),163.9(c-7),161.3(c-5),161.1(c-4’),156.9(c-9),128.6(c-2′,6′),121.0(c-1’),116.0(c-3′,5′),105.3(c-10),103.1(c-3),99.9(c-6),94.8(c-8)。
在对上述数据进行综合分析后,得出化合物c5的分子结构如图16所示,证实化合物c5为芹菜素(cas号:520-36-5)。
6)如图17所示,为化合物c6的nmr氢谱图,其氢谱数据具体为:6.67(1h,s),6.45(1h,d,j=1.8hz),6.19(1h,d,j=1.8hz),7.43(1h,dd,j=8.4,1.2hz),7.43(1h,d,j=2.4hz),6.90(1h,d,j=8.4hz);
如图18所示,为化合物c6的nmr碳谱图,其碳谱数据具体为:181.6(c-4),164.1(c-2),163.9(c-7),161.4(c-5),157.3(c-9),149.7(c-4′),145.7(c-3′),121.5(c-1′),118.9(c-6′),116.0(c-5′),113.4(c-2),103.7(c-l′),102.8(c-3),98.8(c-6),93.8(c-8)。
在对上述数据进行综合分析后,得出化合物c6的分子结构如图19所示,证实化合物c6为木犀草素(cas号:491-70-3)。
7)如图20所示,为化合物c7的nmr氢谱图,其氢谱数据具体为:7.53(1h,d,j=16.0hz,h-3),6.22(1h,d,j=15.6hz,h-2),7.02(1h,d,j=2.0hz,h-2′),6.93(1h,dd,j=8.0hz,h-6′),6.77(1h,d,j=8.0hz,h-5′)。
在对上述数据进行综合分析后,得出化合物c7的分子结构如图21所示,证实化合物c7为咖啡酸(cas号:331-39-5)。
8)如图22所示,为化合物c8的nmr氢谱图,其氢谱数据具体为(cd3od):6.71(1h,s,h-6),6.73(1h,s,h-6),2.62(2h,t,j=7.2hz,h-7),1.80(2h,m,h-8),3.81(2h,t,j=6.5hz,h-9),7.02(1h,d,j=1.8hz,h-2′),7.12(1h,d,j=8.5hz,h-5′),6.92(1h,dd,j=8.5,1.8hz,h-6′),5.54(1h,d,j=5.8hz,h-7′),3.44(1h,m,h-8′),3.81(2h,t,j=6.5,h-9),4.88(1h,d,j=8.0hz,glch-1),3.85(3h,s,3-och3),3.83(3h,s,3’-och3);
如图23所示,为化合物c8的nmr碳谱图,其碳谱数据具体为(cd3od):137.1(c-1),114.1(c-2),145.2(c-3),147.6(c-4),129.5(c-5),117.9(c-6),32.9(c-7),35.8(c-8),62.1(c-9),138.3(c-1′),111.0(c-2′),150.9(c-3′),147.4(c-4′),117.8(c-5′),119.3(c-6′),88.5(c-7′),55.7(c-8′),65.0(c-9′),56.7(3-och3),56.6(3′-och3),102.7(glch-1),74.9(glch-2),78.2(glch-3),71.3(glch-4),77.8(glch-5),62.4(glch-6)。
在对上述数据进行综合分析后,得出化合物c8的分子结构如图24所示,证实化合物c8为urolignoside(cas号:131723-83-6)。
9)如图25所示,为化合物c9的nmr氢谱图,其氢谱数据具体为(dmso):5.45(1h,d,j=7.5hz,h-1"),6.21(1h,d,j=2.1hz,h-6),6.43(1h,d,j=2.1hz,h-8),6.88(2h,d,j=8.9hz,h-3,5′),8.04(2h,d,j=8.9hz,h-2′,6′),12.61(1h,s,5-oh);
如图26所示,为化合物c9的nmr碳谱图,其碳谱数据具体为:61.0(c-6"),70.0(c-4"),74.4(c-2"),76.6(c-3"),77.6(c-5"),93.8(c-8),98.8(c-6),101.0(c-1"),104.2(c-10),115.2(c-3′,5′),121.1(c-1′),131.0(c-2′,6′),133.4(c-3),156.4(c-2),156.5(c-9),160.1(c-4′),161.4(c-5),164.3(c-7),177.6(c-4)。
在对上述数据进行综合分析后,得出化合物c9的分子结构如图27所示,证实化合物c9为紫云英苷(cas号:480-10-4)。
10)如图28所示,为化合物c9的nmr氢谱图,其氢谱数据具体为:12.96(1h,s,5-oh),8.06(2h,d,j=8.6hz,h-2′,6′),7.16(2h,d,j=8.6hz,h-3,5′),6.96(1h,s,h-3),6.80(1h,brs,h-8),6.46(1h,brs,h-6),5.07(1h,d,j=6.7hz,h-1"),4.55(1h,brs,c-1"′),3.86(3h,s,4′-och3),1.07(3h,d,j=5.5hz,h-6"′)。
在对上述数据进行综合分析后,得出化合物c10的分子结构如图29所示,证实化合物c10为蒙花苷(cas号:480-36-4)。
从以上的结果可见,本发明所述的方法成功地将甘木通中的各有效成分逐一分离提纯,而且从氢谱图的峰型可见,分离提纯后得到的物质纯度高,适合于进一步研究或用于制作成药物。
需要指出的是,上述实施例仅是对本发明的进一步说明,而不是限制,本领域技术人员在与本发明技术方案的相当的含义和范围内的任何调整或改变,都应认为是包括在本发明的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!