无定形的B-RAF激酶二聚体抑制剂的制作方法
本发明涉及b-raf激酶二聚体抑制剂1-((1s,1as,6bs)-5-((7-氧代-5,6,7,8-四氢-1,8-二氮杂萘-4-基)氧基)-1a,6b-二氢-1h-环丙并[b]苯并呋喃-1-基)-3-(2,4,5-三氟苯基)脲(以下有时称为化合物1)的一种稳定无定形形式,所述无定形化合物的制备方法和所述无定形化合物的治疗用途。
背景技术:
pct专利申请wo2014/206343a1中公开了第二代b-raf抑制剂,其中包括1-((1s,1as,6bs)-5-((7-氧代-5,6,7,8-四氢-1,8-二氮杂萘-4-基)氧基)-1a,6b-二氢-1h-环丙并[b]苯并呋喃-1-基)-3-(2,4,5-三氟苯基)脲。化合物1的结构如下所示:
作为第二代b-raf抑制剂,化合物1对丝氨酸/苏氨酸激酶的raf家族,特别是braf/craf二聚体具有强效抑制作用。该化合物对mak通道上有突变(包括b-raf突变和k-ras/n-ras突变)的癌症具有靶向治疗作用,是分子靶向治疗剂。相比于第一代b-raf抑制剂,例如威罗菲尼(vemurafenib)和达拉非尼(dabrafenib),化合物1得到了改进。
结晶形式的化合物1作为一种有机合成的游离碱,在水中的溶解度较低,由此口服生物利用度也比较低。
无定形形式是物质多晶型现象中的一种形式,是一种非晶型状态。无定形药物的各种理化性质及临床药效特征常有别于一般的晶型药物。因此,在固体药物的多晶型研究中,对无定形物质的深入探讨同样有着重要意义。一般而言,无定形形式相比于结晶形式具有高分散性与高能状态,因此相比于结晶形式无定形形式具有较高的溶解性与生物利用度。但是,无定形形式由于稳定性较差,其在制药工业领域的应用受到了很大的限制。
本发明的发明人发现了一种化合物1的纯无定形形式,其不同于一般的无定形形式具有相对较长的稳定性,且相比于结晶形式具有相对较高的生物利用度。
技术实现要素:
本发明提供一种化合物1的纯无定形形式,所述无定形形式在14天的测试期间没有发生晶形转化,即在14天的测试期间内依然没有显示任何衍射峰;而且所述无定形形式相比于结晶形式具有相对较高的生物利用度。这表明,本发明的无定形形式具备药物制备的潜在用途。
第一方面,本发明提供化合物1的纯无定形形式,其特征在于,所述纯无定形形式的x-射线粉末衍射图无衍射峰,如图7所示。
优选地,本发明的纯无定形形式具有如图11所示的1h-nmr谱。
优选地,本发明的纯无定形形式的玻璃态转变温度在约135至143℃之间,更优选为约138.3℃。
优选地,本发明的纯无定形形式的粒度为在约60至约80μm之间的d90,在约2至约6μm之间的d50,在约1至约2μm之间的d10;更优选地d90为约69.9μm、d50为约3.5μm,以及d10为约1.4μm。
第二方面,本发明提供制备化合物1的纯无定形形式的方法,所述方法包括:对化合物1的结晶形式在极性溶剂中的溶液进行喷雾干燥,得到粉末状物质。
优选地,所述极性溶剂包括醚类、羧酸酯类、腈类、酮类、酰胺类、砜类、亚砜类或卤代烃类。更优选地,所述极性溶剂包括但并不限于,乙酸、丙酮、乙腈、苯、氯仿、四氯化碳、二氯甲烷、二甲基亚砜、1,4-二氧六环、乙醇、乙酸乙酯、丁醇、叔丁醇、n,n-二甲基乙酰胺、n,n-二甲基甲酰胺、甲酰胺、蚁酸、庚烷、己烷、异丙醇、甲醇、甲基乙基酮、l-甲基-2-吡咯烷酮、均三甲苯、硝基甲烷、聚乙二醇、丙醇、2-丙酮、吡啶、四氢呋喃、甲苯、二甲苯、它们的混合物等等。优选地,所述极性溶剂为卤代烃类/酰胺类的混合物,例如dcm/meoh的混合物。
优选地,所述喷雾干燥通过喷雾干燥器进行。优选地,所述喷雾干燥器的入口温度设定为约50至70℃,所述喷雾干燥器的出口温度设定为约25至45℃;更优选地,所述喷雾干燥器的入口温度设定为约60℃,所述喷雾干燥器的出口温度设定为约35℃。
优选地,所述化合物1的结晶形式是结晶形式a。在一个实施方案中,所述结晶形式a的特征在于包含至少三个、四个、五个或六个具有独立选自下组2θ°值的衍射峰的x射线粉末衍射图:4.7±0.2、9.4±0.2、13.6±0.2、14.0±0.2、14.9±0.2和15.6±0.2°。优选地,所述结晶形式a的特征在于包含至少三个、四个、五个或六个具有独立选自下组2θ°值的衍射峰的x射线粉末衍射图:4.7±0.2、9.4±0.2、13.6±0.2、14.0±0.2、14.9±0.2、15.6±0.2、21.2±0.2、24.3±0.2、24.7±0.2、25.1±0.2和29.1±0.2°。更优选地,所述结晶形式a的特征在于包含具有独立选自下组2θ°值的衍射峰的x射线粉末衍射图:4.7±0.2、9.4±0.2、10.2±0.2、13.6±0.2、14.0±0.2、14.9±0.2、15.6±0.2、17.2±0.2、17.4±0.2、18.7±0.2、20.0±0.2、20.4±0.2、21.2±0.2、22.3±0.2、24.3±0.2、24.7±0.2、25.1±0.2、25.5±0.2、26.8±0.2、27.4±0.2、27.8±0.2、28.6±0.2、29.1±0.2、30.2±0.2、31.8±0.2、32.0±0.2、33.1±0.2、34.1±0.2和34.6±0.2°。在一个优选的实施方案中,所述结晶形式a的特征在于基本上如图1中所示的x射线粉末衍射图。
优选地,所述化合物1的结晶形式是单晶a**。
在一个实施方案中,所述单晶形式a**的晶胞参数
在一个实施方案中,所述单晶形式a**的空间群为单斜(晶系)p21。
在一个优选的实施方案中,所述结晶形式a是通过商业规模方法制备的。
第三方面,本申请公开了治疗或预防对受试者中raf激酶的抑制有响应的疾病或病症的方法,包括给予所述受试者治疗有效量的化合物1,其中化合物1呈本申请公开的纯无定形形式。
在一个实施方案中,所述疾病或病症是选自下组的癌症:脑癌、肺癌、肾癌、骨癌、肝癌、膀胱癌、乳腺癌、头颈癌、卵巢癌、黑素瘤、皮肤癌、肾上腺癌、宫颈癌、淋巴瘤或甲状腺肿瘤及其并发症。
在另一个实施方案中,所述疾病是选自下组的braf(v600e或非v600e)或nras或kras突变癌症:脑癌、肺癌、肾癌、骨癌、肝癌、膀胱癌、乳腺癌、头颈癌、卵巢癌、黑素瘤、皮肤癌、肾上腺癌、宫颈癌、淋巴瘤或甲状腺肿瘤及其并发症。
在另一个实施方案中,化合物1的给药剂量为1-200mg/天,并且给药频率为每天一至三次。
在另一个实施方案中,化合物1的给药剂量为2.5-100mg/天,并且给药频率为每天一至三次。
在另一个实施方案中,化合物1的给药剂量为5-50mg/天,并且给药频率为每天一次。
在一个实施方案中,受试者是大鼠、犬或人。
第四方面,本申请公开了药物组合物,其包含治疗有效量的化合物1,所述化合物1呈本申请公开的纯无定形形式。其中,活性化合物,即化合物1可占药物组合物的1-99%(重量),优选1-50%(重量),更优选1-30%(重量),或最优先1-20%(重量)。
药物组合物可以如下形式给药:以诸如胶囊、片剂、丸剂、粉末形式口服给药,持续释放注射剂(诸如无菌溶液、悬浮液或乳液)形式给药;通过局部治疗形式,诸如糊剂、乳霜或软膏;或通过栓剂诸如栓剂形式。药物组合物可以是适合于精确剂量应用的单位剂型。
合适的药物载体包括水、各种有机溶剂和各种惰性稀释剂或填充剂。如果需要,药物组合物可含有各种添加剂,诸如香料、粘合剂和赋形剂。对于口服给药,片剂和胶囊可含有各种赋形剂,诸如柠檬酸;各种崩解剂,诸如淀粉、海藻酸和一些硅酸盐;以及各种粘合剂,诸如蔗糖、明胶和阿拉伯胶。此外,包括硬脂酸镁和滑石填料的润滑剂通常用于片剂的生产。相同类型的固体组分也可用于配制软和硬明胶胶囊。当口服给药需要含水悬浮液时,活性化合物可与各种甜味剂或调味剂、颜料或染料组合混合。如果需要,可以使用各种乳化剂或产生悬浮液;可以使用稀释剂诸如水、乙醇、丙二醇、甘油或它们的组合。
上述药物组合物优选口服给药。
上述药物组合物优选呈胶囊或片剂形式。
附图说明
图1显示化合物1的一种结晶形式(形式a)的x射线衍射图(从异丙醇/水中结晶)。
图2显示化合物1的另一种结晶形式(形式a*)的x射线衍射图。
图3显示化合物1的单晶(形式a**)(通过从乙酸乙酯/庚烷中结晶获得的单晶)的绝对结构。
图4显示化合物1的单晶(形式a**)的晶体堆积。
图5说明了化合物1的单晶(形式a**)的氢键。
图6显示使用mercury软件计算的化合物1的单晶(形式a**)的理论xrpd图。
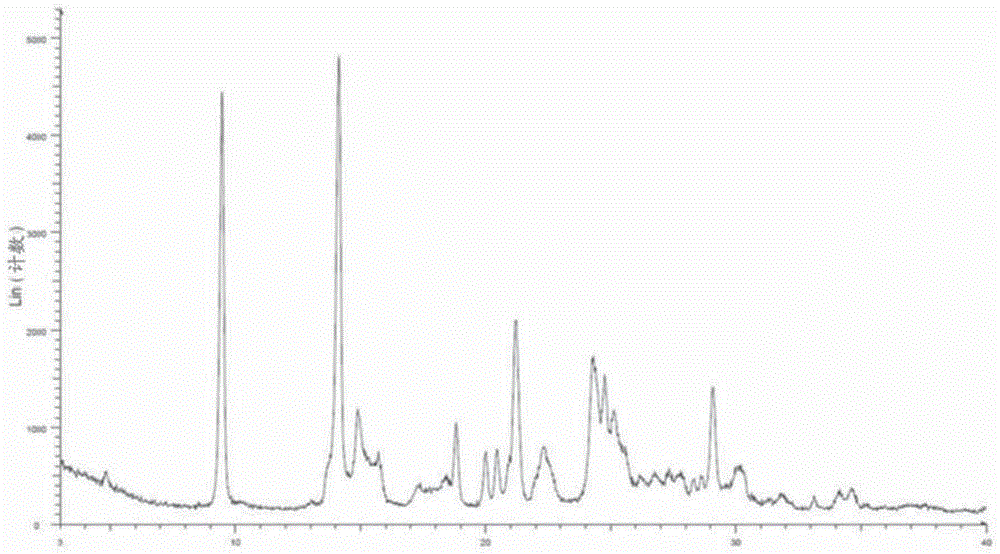
图7显示化合物1的纯无定形形式(形式b)的x射线衍射图。
图8显示化合物1的结晶形式(形式a)的1h-nmr谱图。
图9显示化合物1的结晶形式(形式a)的l3c-nmr谱图。
图10显示化合物的结晶形式a的dvs吸湿性(即水分吸收)。
图11显示化合物1纯无定形形式(形式b)的1h-nmr谱。
图12显示化合物1纯无定形形式(形式b)的重叠。
具体实施方式
定义和一般术语
除非另有说明,本发明使用的所有技术和科学术语与本发明所属领域的普通技术人员所通常理解的具有相同含义。本发明涉及的所有专利和公开出版物通过引用方式整体并入本发明。尽管在本发明的实践或者测试中可以使用与本发明所述相似或者相同的任何方法和物质,但是本发明中描述的是优选的方法、设备和物质。
“晶型”或“结晶形式”是指具有高度规则化学结构的固体,包括,但不限于,单组分或者多组分晶体,和/或化合物的多晶型物、溶剂化物、水合物、包合物、共晶、盐、盐的溶剂化物、盐的水合物。物质的结晶形式可通过本领域已知的多种方法得到。这些方法包括但不限于,熔体结晶、熔体冷却、溶剂结晶、在限定的空间中结晶,例如在纳米孔或者毛细管中,在表面或者模板上结晶,例如在聚合物上,在添加剂如共结晶反分子的存在下结晶、去溶剂、脱水、快速蒸发、快速冷却、缓慢冷却、蒸气扩散、升华、反应结晶、反溶剂添加、研磨和溶剂滴研磨等。
“无定形”或“无定形形式”是指物质的质点(分子、原子、离子)在三维空间排列无周期性时形成的物质,其特征是具有漫射的不具尖峰的x射线粉末衍射图。无定形是固体物质的一种特殊的物理形式,其局部有序的结构特征,提示其与晶型物质有着千丝万缕的联系。物质的无定形形式可通过本领域已知的多种方法得到。这些方法包括,但不限于,骤冷法、反溶剂絮凝法、球磨法、喷雾干燥法、冷冻干燥法、湿法制粒法和固体分散体技术等等。本发明所述的“纯无定形形式”是指仅由化合物1组成的无定形形式,而没有任何其它赋型剂。
“极性溶剂”是指含有羟基或羰基等极性基团的溶剂,其极性强,介电常数大。适于本发明目的的极性溶剂包括但并不限于乙酸、丙酮、乙腈、苯、氯仿、四氯化碳、二氯甲烷、二甲基亚砜、1,4-二氧六环、乙醇、乙酸乙酯、丁醇、叔丁醇、n,n-二甲基乙酰胺、n,n-二甲基甲酰胺、甲酰胺、蚁酸、庚烷、己烷、异丙醇、甲醇、甲基乙基酮、l-甲基-2-吡咯烷酮、均三甲苯、硝基甲烷、聚乙二醇、丙醇、2-丙酮、吡啶、四氢呋喃、甲苯、二甲苯、以及它们中两者、三者或更多的混合物等等。
晶型或无定形可以通过多种技术手段进行确定,例如x射线粉末衍射(xrpd)、红外吸收光谱法(ir)、熔点法、差示扫描量热法(dsc)、热重分析法(tga)、核磁共振法、拉曼光谱、x射线单晶衍射、溶解量热法、扫描电子显微镜(sem)、定量分析、溶解度和溶解速度等等。
x射线粉末衍射(xrpd)可检测晶型的变化、结晶度、晶构状态等信息,是鉴别晶型的常用手段。xrpd图的峰位置主要取决于晶型的结构,对实验细节相对不敏感,而其相对峰高取决于与样品制备和仪器几何形状有关的许多因素。因此,在一些实施例中,本发明的晶型的特征在于具有某些峰位置的xrpd图,其基本上如本发明附图中提供的xrpd图所示。同时,xrpd图的2θ的量度可以有实验误差,不同仪器以及不同样品之间,xrpd图的2θ的量度可能会略有差别,因此所述2θ的数值不能视为绝对的。根据本发明试验所用仪器状况,衍射峰存在±0.2°的误差容限。
差示扫描量热(dsc)是通过程序控制,不断加热或降温,以测量样品与惰性参比物(常用α-al2o3)之间的能量差随温度变化的技术。dsc曲线的熔化峰高取决于与样品制备和仪器几何形状有关的许多因素,而峰位置对实验细节相对不敏感。因此,在一些实施例中,本发明所述晶型或无定形的特征在于具有特征峰位置的dsc图,其基本上如本发明附图中提供的dsc图所示。同时,dsc图谱可以有实验误差,不同仪器以及不同样品之间,dsc图谱的峰位置和峰值可能会略有差别,因此所述dsc吸热峰的峰位置或峰值的数值不能视为绝对的。根据本发明试验所用仪器状况,熔化峰存在±3℃的误差容限。
玻璃态转变是指非晶态物质在高弹态和玻璃态之间的转变,是该物质的固有性质;它所对应的转变温度为玻璃化转变温度(tg),是非晶态物质的一个重要物理性质。玻璃化转变是与分子运动有关的现象。因此,玻璃化转变温度(tg)主要取决于物质的结构,而对实验细节等相对不敏感。在一些实施方案中,本发明所述无定形的玻璃化转变温度(tg)通过差示扫描量热法(dsc)测定,其特征在于具有138.3℃的玻璃化转变温度。根据本发明试验所用仪器状况,玻璃化转变温度存在±3℃的误差容限。
差示扫描量热(dsc)还可用于检测分析晶型是否有转晶或混晶现象。
化学组成相同的固体,在不同的热力学条件下,常会形成晶体结构不同的同质异构体,或称为变体,这种现象称为同质多晶或同质多相现象。当温度和压力条件变化时,变体之间会发生相互转变,此现象称为晶型转变。由于晶型转变,晶体的力学、电学、磁学等性能会发生巨大的变化。当晶型转变的温度在可测范围内时,在差示扫描量热(dsc)图上可观察到这一转变过程,其特征在于,dsc图具有反映这一转变过程的放热峰,且同时具有两个或多个吸热峰,分别为转变前后的不同晶型的特征吸热峰。
热重分析(tga)是通过程序控制,测定物质的质量随温度变化的技术,适用于检查晶体中溶剂的丧失或样品升华、分解的过程,可推测晶体中含结晶水或结晶溶剂的情况。tga曲线显示的质量变化取决于样品制备和仪器等许多因素;不同仪器以及不同样品之间,tga检测的质量变化略有差别。根据本发明试验所用的仪器状况,质量变化存在±0.1%的误差容限。
在本发明的上下文中,x-射线粉末衍射图中的2θ值均以度(°)为单位。
术语“基本上如图所示”是指x-射线粉末衍射图或dsc图至少50%,或至少60%,或至少70%,或至少80%,或至少90%,或至少95%,或至少99%的峰显示在其图中。
当提及谱图或/和出现在图中的数据时,“峰”指本领域技术人员能够识别的不会归属于背景噪音的一个特征。
“相对强度”是指x-射线粉末衍射图(xrpd)的所有衍射峰中第一强峰的强度为100%时,其它峰的强度与第一强峰的强度的比值。
除非另有说明,否则本申请所用的术语“约”表示数量(例如,温度、ph、体积等)可在±10%内变化,优选在±5%内变化。
在一个实施方案中,本发明结晶形式根据下述方案1合成。值得注意的是,本申请公开的方法特别适用于以高质量、高产率可再现的、商业规模的制造化合物1或其结晶形式a。
方案1
以下合成方法、具体实施例和功效测试进一步描述了本发明,但是它们不应被解释成以任何方式限定或限制本发明的范围。
实施例
以下实施例旨在是示例性的。尽管已经做出努力以确保关于所使用的数字(例如,量、温度等)的准确性,但是应该考虑一些实验误差和偏差。除非另有说明,否则温度为摄氏度。试剂购自商业供应商,诸如sigma-aldrich、alfaaesar或tci,除非另有说明,否则无需进一步纯化即可使用。
除非另有说明,否则下述反应在氮气或氩气的正压力下或在无水溶剂中用干燥管进行;反应烧瓶配有橡胶隔片,用于通过注射器引入底物和试剂;将玻璃器皿烘箱干燥和/或加热干燥。
除非另有说明,否则柱色谱纯化在具有硅胶柱的biotage系统(制造商:dyaxcorporation)上或在二氧化硅seppak柱(waters)上进行,或者在使用预填充硅胶管柱的teledyneiscocombiflash纯化系统上进行。
在400mhz操作的varian仪器上记录1hnmr谱和13cnmr谱。
使用brukerapex-iiccd衍射仪(cukα辐射,
在以下实施例中,可以使用以下缩写:
acoh乙酸
acn乙腈
api活性药物成分
aq水性或水溶液
brine饱和氯化钠水溶液
bn苄基
bnbr苄基溴
ch2cl2二氯甲烷
dman,n-二甲基乙酰胺
dmfn,n-二甲基甲酰胺
dppf1,1’-双(二苯基膦基)二茂铁
dbu1,8-二氮杂双环[5.4.0]十一-7-烯
dcm二氯甲烷
diea或dipean,n-二异丙基乙基胺
dmap4-n,n-二甲基氨基吡啶
dmfn,n-二甲基甲酰胺
dmso二甲基亚砜
etoac乙酸乙酯
etoh乙醇
et2o或ether乙醚
g克
h或hr小时
hatuo-(7-氮杂苯并三唑-1-基)-n,n,n',n'-四甲基脲鎓六氟磷酸盐
hcl盐酸
hplc高效液相色谱
hpmcas乙酸琥珀酸羟丙基甲基纤维素
ipa或i-proh2-丙醇或异丙醇
mg毫克
ml毫升
mmol毫摩尔
mecn乙腈
meoh甲醇
min分钟
ms或ms质谱
na2so4硫酸钠
pe石油醚
ppa多聚磷酸
rt保留时间
rt或rt室温
tbaf四丁基氟化铵
tbscl叔丁基二甲基氯硅烷
tea三乙醇胺
tfa三氟乙酸
thf四氢呋喃
tlc薄层色谱
tmscl三甲基氯硅烷
μl微升
xrpdx-射线粉末衍射
实施例1
化合物1结晶形式a的制备
步骤1:intq-1的合成
将1,4-二噁烷(1.5体积)添加至2l的4颈圆底烧瓶中,将烧瓶抽空并用氮气冲洗三次。然后将pd(oac)2(2wt%,0.50kg)和xantphos(9wt%,2.25kg)添加至烧瓶中,将烧瓶抽空并用氮气冲洗三次。在氮气气氛下将混合物在室温搅拌0.5~1小时。将naoh(12.25kg,1.6当量)、h2o(1体积,25l)和1,4-二噁烷(8体积,200l)添加至20l反应器中。搅拌混合物直至澄清,然后将sm3(26.75kg,1.2当量)添加至混合物中。在氮气气氛下将催化剂溶液转移到上述反应器中。然后将sm1(25.00kg,1.0当量)逐滴添加至反应器中。将体系加热至65±5℃并保持在65±5℃至少5小时。hplc用于监测反应直至sm1的含量不超过1.0%。将反应混合物冷却至30±5℃,然后过滤,滤饼用1,4-二噁烷(1.0体积)洗涤。将h2o(4体积)添加至滤液中并浓缩至5体积。然后将h2o(2体积)添加至残余物中并浓缩至5体积。将残余物冷却至室温并过滤。将滤饼用h2o(2体积)洗涤。然后将滤饼用ipa(2体积)在25±5℃浆化3小时。过滤混合物,滤饼用ipa(0.5体积)洗涤。将固体在减压下在烘箱中干燥。
步骤2和3:intq-3的合成
将thf(25体积)和intq-1(16.00kg,1.0当量)添加至反应器中。搅拌混合物并冷却至-80~-70℃。然后在-80~-70℃向混合物中逐滴添加n-buli(正己烷溶液,2.5m,51.20kg,2.5当量)。在-80~-70℃反应1-2小时后,通过tlc监测反应。然后在-80~-70℃将dmf(9.92kg,1.8当量)的thf(1.4体积)溶液逐滴添加至反应体系中。在-80~-70℃反应1-2小时后,通过tlc监测反应。在-80~-70℃将acoh于thf(1.4体积)中的溶液逐滴添加至混合物中,以将ph值调节至6~7。然后在-80~-70℃将tea(8.00kg,1.05当量)添加至反应中。向反应混合物中逐滴添加甲基三苯基亚正膦基乙酸酯(26.4kg,1.05当量)在dcm(19体积)中的溶液。将混合物在-80~-70℃搅拌10小时,然后通过tlc监测反应。将h2o(10.5体积)和柠檬酸(32.00kg,2.1当量)加入另一反应器中。搅拌混合物使其溶解并冷却至0~5℃。将温度冷却至-20℃并将溶液转移到上述3l的4颈圆底烧瓶中。然后将混合物在20℃以下搅拌1小时,确认ph值在4~7之间。分离有机层并用25%nacl(17体积)洗涤。然后将有机相浓缩至5体积并将etoac(17体积)加入混合物中并浓缩至5体积。将etoac(17体积)加入混合物中并浓缩至5体积。该溶液直接用于下一步骤。
步骤4:intq-4的合成
将intq-3于etoac的溶液加入反应器中。搅拌溶液并冷却至-5~5℃。在-5~5℃将hcl加入混合物中保持2小时。然后将混合物加热至20~30℃。在反应5小时后,每2小时使用hplc监测反应,直至intq-3的含量小于0.5%。将反应混合物浓缩至10体积并冷却至0~5℃。将残余物在0~5℃搅拌1小时。过滤混合物,将滤饼加入h2o(15体积)中。将混合物在20~30℃搅拌2小时。过滤混合物,滤饼用h2o(3体积)洗涤。然后将滤液转移到另一个反应器中并将na2co3加入混合物中以将ph值调节至8~9。然后过滤混合物,滤饼用h2o(4体积)洗涤。在真空烘箱中干燥后,得到20.73kg(产率:69.0%,纯度:95.0%)的intq-4。
步骤5:intq-5的合成
将intq-4(10.40kg,1.0当量)、pd/c(15%wt,1.25kg)和thf(11体积)加入反应器中。搅拌混合物并加热至30~35℃。加入氢气至10atm的压力。在反应15小时后,每2小时使用hplc监测反应,直至intq-4的含量小于0.5%。将反应混合物冷却至20-30℃并通过硅藻土(0.2wt)过滤。将滤饼用thf(2体积)洗涤。将滤液浓缩至3体积并将etoh(6体积)添加至混合物中。将溶液浓缩至3体积并将etoh(6体积)加入混合物中。将混合物浓缩至3体积并直接用于下一步骤。
步骤6:bgb-intq-6的合成
将intq-5(来自前一步骤)于etoh(3体积)中的溶液、etoh(7体积)和et3n(22%wt,2.29kg)加入反应器中。将溶液加热至70~80℃。反应15小时后,每隔2小时用hplc监测反应,直至intq-5的含量小于1.0%。将反应混合物冷却至30~40℃并浓缩至5体积。将混合物冷却至-5~0℃并搅拌2小时。过滤混合物,滤饼用etoh(1体积)洗涤。在45±5℃的烘箱中干燥后,得到7.58kg(产率:87.1%,纯度:99.5%)的intq-6。
步骤7:intq-7的合成
将氢氧化钾(49.9kg,1.7当量)添加至4-甲氧基苯酚(65kg,1.0当量)于dmso(65l,1体积)中的溶液中。将系统加热至120℃。逐滴添加溴乙醛二乙缩醛(123.8kg,1.2当量),同时保持温度在120~140℃。通过hplc监测反应完成后将反应混合物冷却至20~40℃。将正庚烷(2体积)和水(2体积)加入到反应混合物中。将混合物通过硅藻土(0.2wt)过滤,并将滤饼用正庚烷(0.5体积)洗涤。将滤液静置至少30分钟。分离有机层,水层用正庚烷(2体积)萃取。将合并的有机层用2nnaoh水溶液(2体积)洗涤。将有机层用15%nacl水溶液(2体积)洗涤两次。将有机层浓缩至3体积。添加甲苯(3体积)并继续浓缩至3体积。intq-7的甲苯溶液直接用于下一步骤。
步骤8:intq-8的合成
将amberlyst-15(3.8kg,0.1wt)添加至甲苯(760l,20体积)中。在n2保护下将系统加热至110℃。逐滴添加intq-7(38kg/批,3批,1.0当量)于甲苯中的溶液,同时保持温度在105~110℃。反应1小时后,将反应体系在105~110℃的恒定压力下浓缩至17体积。将甲苯(3倍体积)加入系统中。通过hplc监测反应完成后将反应混合物冷却至20~40℃。将混合物通过硅藻土(0.1wt)过滤,滤饼用甲苯(0.5体积)洗涤。滤液用2nnaoh水溶液(2体积)洗涤。有机层用20%nacl水溶液(2体积)洗涤两次。将有机层浓缩至2体积。将粗产物在110℃以下蒸馏,得到intq-8,其为灰白色固体(43kg,收率=61.2%,纯度≥98.0%)。
步骤9:intq-9的合成
将1-十二烷硫醇(147.0kg,3.5当量)添加至intq-8(43kg,1.0当量)于nmp(260l,6体积)中的溶液中。将体系加热至75±5℃。分批加入乙醇钠(69.0kg,3.5当量),同时保持温度低于120℃。将反应混合物加热至130±5℃。每小时对混合物取样用于hplc,直至在130±5℃反应16小时后ph-bei-bgb-3289-intq-8的含量≤3.0%。将反应混合物冷却至60±5℃,然后向混合物中加入8体积水。将反应混合物冷却至25±5℃,然后向混合物中加入3体积石油醚。将混合物搅拌至少30分钟并静置至少30分钟,分离。有机相是临时存储的。用6nhcl将水相调节至ph=1~2。分别用5体积和3体积乙酸乙酯萃取水相。将残余物水溶液与临时有机相合并,然后加入4体积乙醇和4体积石油醚。将混合物搅拌至少30分钟并静置至少30分钟,然后分离。用6nhcl将水相调节至ph=1~2。用5倍体积乙酸乙酯萃取水相。合并乙酸乙酯的有机相,并在低于50℃的压力下浓缩至3体积。向残余物中加入5体积正庚烷,并用5%naoh将混合物调节至ph=9~10。将混合物搅拌至少30分钟并静置至少30分钟,分离。用6nhcl将水相调节至ph=1~2。分别用5体积和3体积乙酸乙酯萃取水相。然后合并乙酸乙酯的有机相,用6体积10%h2o2和浓hcl(0.15wt)洗涤。然后用6体积5%h2o2和浓hcl(0.15wt)洗涤有机相。用4体积5%na2so3洗涤有机层。用3体积盐水洗涤有机层三次。将有机层浓缩至3体积。添加二氯甲烷(5体积)并继续浓缩至无明显级分(fraction)。intq-9的粗产物直接用于下一步骤。
步骤10:intq-10的合成
将et3n(48.2kg,2.0当量)添加至低于40℃的intq-9(32kg,1.0当量)于二氯甲烷(10体积)中的溶液中。将混合物冷却至-5±5℃。逐滴添加于二氯甲烷(1体积)中的tmscl(1.3当量),同时保持温度在-5±5℃。每小时对混合物取样用于气相色谱,直至在-5±5℃反应1小时后intq-9的含量≤2.0%。将混合物在低于40℃的压力下浓缩至3体积。向残余物中加入15体积的正己烷,并将混合物搅拌至少30分钟。过滤混合物,在低于40℃的压力下浓缩滤液至没有明显的级分。将粗产物在120℃以下蒸馏,得到intq-10,其为浅黄色油状物(40kg,收率=81.4%,纯度≥97.5%)。
步骤11:intq-11的合成
将于二氯甲烷(5体积)中的intq-10(20kg/批,2批,1.0当量)与cui(0.1wt)在25±5℃浆化2~3小时。在n2气氛下,在20~30℃,将三氟甲磺酸亚铜(i)(与甲苯的2:1络合物,0.11%wt)和(s,s)-2,2-双(4-苯基-2-噁唑啉-2-基)丙烷(0.15%wt)于二氯甲烷(4体积)中搅拌2~3小时。通过小孔过滤器加入intq-10于二氯甲烷中的溶液,在20~30℃在15~25小时内缓慢逐滴添加重氮乙酸乙酯(2.0当量)于二氯甲烷(10体积)中的溶液。将混合物在20~30℃搅拌30~60分钟,在20~30℃用4体积0.05n乙二胺四乙酸二钠二水合物洗涤混合物三次。用3体积25%nacl水溶液洗涤有机部分两次。将有机部分在低于35℃的真空下浓缩,直至系统不超过3体积。intq-11的粗产物直接用于下一步骤。
步骤12和13:intq-13的合成
步骤12:将intq-11的粗产物溶于甲醇(3体积)中,将38%hcl/etoh(0.1体积)添加至混合物中并在20~30℃搅拌2~3小时。向混合物中逐滴添加et3n以调节ph=7。将混合物在压力下浓缩至2体积。加入乙酸乙酯(2体积)并继续在压力下浓缩至2体积。加入正庚烷(2体积)并继续在压力下浓缩至2体积。加入二氯甲烷(2体积)使该物质完全溶解。通过硅胶色谱法纯化残余物(用etoac:pe=1:5洗脱,总共约100体积),得到intq-12,其为黄色固体。
步骤13:将intq-12加入etoac(1.5体积)和正庚烷(20体积)中,将混合物加热至75~85℃直至澄清。将澄清溶液在75~85℃搅拌1小时,然后逐渐冷却至15~20℃。过滤混合物并用正庚烷(2体积)洗涤,得到产物。将湿产物在55±5℃干燥至少16小时,得到intq-13,其为浅黄色至灰白色固体。
步骤14和15:intq-15的合成
步骤14:将intq-13(16kg,1.0当量)和intq-6(12.7kg,1.05当量)添加至dmf(5体积)中。将体系加热至55±5℃。添加碳酸铯(29.6kg,1.25当量)。将反应混合物加热至110±5℃。每小时对混合物取样用于hplc,直至在110±5℃反应2小时后intq-13的含量≤0.5%。将反应混合物冷却至30±5℃,然后用乙酸(5wt)在30±5℃调节至ph=6。在25±5℃将水(30体积)添加至混合物中。将混合物搅拌1~2小时并过滤,得到湿产物。将湿产物与水(5体积)再浆化。滤饼直接用于下一步骤。
步骤15:将intq-14的湿产物添加至1nnaoh(10体积)和thf(20体积)的混合物中。将体系在25±5℃搅拌。每小时取样用于hplc,直至在25±5℃反应4小时后intq-14的含量≤0.5%。用4nhcl在25±5℃将体系调节至ph=4~5并搅拌1小时。在低于50℃的压力下将体系浓缩至8体积,然后过滤,得到湿产物。将湿产物与thf(10体积)再浆化。将混合物搅拌1~2小时并过滤,得到湿产物。将湿产物在55±5℃干燥至少30小时,得到intq-15,其为浅棕色至灰白色固体。
步骤16和17和18:intq-18的合成
将反应器抽真空至≤-0.08mpa,然后充入惰性氮气。将1,4-二噁烷(10.0体积),intq-15(3.6kg,1.0当量)添加至反应器中。将混合物在低于50℃浓缩至6.0~6.5体积,并对混合物取样以获得水的含量。将et3n(1.1当量)加入反应器中。将混合物加热至30±5℃,并将dppa(1.1当量)逐滴添加至反应器中。在30±5℃反应2小时后,将混合物取样用于hplc分析,直至intq-15的含量≤1.0%。获得了intq-16的溶液。
将反应器抽真空至≤-0.08mpa,然后充入惰性氮气。将t-buoh(20.0体积)、(boc)2o(0.5当量)和dmap(0.02当量)加入反应器中。将混合物加热至85±5℃,搅拌2~3小时,并对混合物中的水含量进行取样。标准是kf≤0.01%。将intq-16的溶液在85±5℃滴加到上述t-buoh体系的反应器中(持续至少3小时)。在85±5℃反应2小时后,将混合物取样用于hplc分析,直至intq-16的含量≤1.0%。然后将混合物冷却至低于50℃,并在50℃以下浓缩至3.0~4.0体积。
将dcm(10.0体积×2)加入到残余物中,并将混合物在50℃以下浓缩至3.0~4.0体积。将dcm(10.0体积)加入到残余物中。然后将1wt%naoh水溶液(20.0体积)加入反应器中并在25±5℃搅拌至少1小时。将混合物通过硅藻土过滤,然后分离。用水(5.0体积)洗涤有机相并分离。用25wt%盐水(5.0体积)进一步洗涤有机相,用硅胶垫过滤分离以除去部分杂质。将有机相在40℃以下浓缩至6.0~7.0体积。加入dcm至7.0体积。然后将混合物冷却至不超过15℃,并在不高于15℃的温度将盐酸(1.2体积)逐滴添加至反应器中。在15±5℃反应3小时后,将混合物取样用于hplc分析,直至intq-17的含量≤4.0%。将混合物加热至25±5℃,向反应器中添加水(3.0体积)。
intq-16:1hnmr(400mhz,dmso-d6)δ10.48(s,1h),7.95(d,j=5.6hz,1h),7.34(d,j=2.4hz,1h),7.05(d,j=8.4hz,1h),7.00(dd,j=8.8,2.4hz,1h),6.25(d,j=5.6hz,1h),5.42(d,j=5.2hz,1h),3.56(dd,j=5.2,2.8hz,1h),2.92(t,j=7.6hz,2h),2.54(d,j=8.0hz,2h),1.51(d,j=3.2hz,1h).ms:m/e364(m+1)+.
intq-17:1hnmr(400mhz,dmso-d6)δ10.49(s,1h),7.94(d,j=6.0hz,1h),7.34(s,1h),7.18(s,1h),6.96–6.83(m,2h),6.22(d,j=5.6hz,1h),4.86(d,j=5.6hz,1h),2.92(t,j=7.6hz,2h),2.86(d,j=4.8hz,1h),2.54(t,j=7.6hz,2h),2.12(s,1h),1.39(s,9h).ms:m/e410(m+1)+.
ph调节过程:将4wt%naoh水溶液逐滴添加至反应器中以将ph值调节至2.7~3.1。如果ph>3.1,则加入盐酸(0.2体积),然后将4wt%naoh水溶液逐滴添加至反应器中,以将ph值调节到2.7~3.1(精密ph试纸,范围2.7~4.7);分离混合物,收集乳液相作为水相。将混合物通过硅藻土过滤,并将所得水相用dcm(2.0体积)洗涤一次。向反应器中的剩余水相中加入dcm(6.0体积)和etoh(5.0体积)。将10.0wt%na2co3溶液逐滴添加至反应中,以在25±5℃将ph值调节至8~9。将混合物搅拌10~15分钟并静置10~15分钟。分离混合物,水相用dcm(4.0体积)萃取2次。合并有机相,用水(2.0体积)洗涤,分离,有机相用25wt%盐水(5.0体积)洗涤一次。将有机相在45℃以下浓缩至3.0~4.0体积,然后将正庚烷(4.0体积)加入到残余物中。将混合物在45℃以下浓缩至3.0~4.0体积,然后将正庚烷(4.0体积)加入到残余物中。将混合物在45℃以下浓缩至3.0~4.0体积。将残余物冷却至25±5℃,然后离心,用正庚烷(2.0体积)洗涤固体。将滤饼转移到真空烘箱中,在45±5℃(箱温)干燥4小时后,将混合物取样用于干燥失重(lod),直至lod≤1.0%。报道了intq-18(2.25kg)的纯度。将产品包装在双ldpe塑料袋中并在2~30℃储存。
intq-18:1hnmr(400mhz,dmso-d6)δ10.81(s,1h),8.87(s,3h),8.05(d,j=6.0hz,1h),7.33(t,j=1.2hz,1h),7.07–6.95(m,2h),6.34(d,j=6.0hz,1h),5.24(d,j=6.0hz,1h),3.32(dd,j=6.0,2.0hz,1h),2.97(t,j=7.6hz,2h),2.59(t,j=7.6hz,2h),2.46(s,1h).ms:m/e310(m+1)+.
步骤19:intq-19的合成
将反应器抽真空至≤-0.08mpa,然后充入惰性氮气。将thf(6.0体积)、h2o(3.0体积)、2,4,5-三氟苯胺(1.0当量)、nahco3(1.2当量)加入反应器中。将混合物冷却至0℃,在0±5℃缓慢添加氯甲酸苯酯。将混合物搅拌至少2小时。对混合物取样用于lcms,直至2,4,5-三氟苯胺≤0.2%。然后添加ea(15.0体积)。用h2o(5.0体积)洗涤有机相,然后用5wt%hcl水溶液(5.0体积)洗涤2次,用饱和nacl(5.0体积)洗涤2次。将有机相在低于45℃浓缩至10.0体积。将正庚烷(10.0体积)加入残余物中。将混合物浓缩至10.0体积,然后将正庚烷(10.0体积)加入到残余物中。将混合物浓缩至10.0体积并离心,用正庚烷(2.0体积)洗涤固体。将滤饼取样用于lcms分析,其中intq-19的标准>99%。然后将滤饼转移到真空烘箱中并在35±5℃(箱温)干燥10小时后取样lod,直至lod≤2.0%。报道了intq-19的纯度。将产品包装在双ldpe塑料袋中并在2-30℃储存。
intq-19:1hnmr(400mhz,dmso-d6)δ10.20(s,1h),7.82(dt,j=12.0,8.0hz,1h),7.66(td,j=10.8,7.6hz,1h),7.44(t,j=7.6hz,2h),7.33–7.20(m,3h).
步骤20:化合物1的结晶形式(形式a)的合成
将反应器抽真空至≤-0.08mpa,然后充入惰性氮气。将dmso(9.0体积)、intq-18(1.63kg,1.0当量)和n-甲基吗啉(nmm,1.0当量)加入反应器中。将混合物在20±5℃搅拌至少0.5小时。将intq-19(1.27kg,0.9当量)在20±5℃加入反应器中。在20±5℃反应3小时后,将混合物取样用于hplc分析,直至intq-19的含量≤0.3%。反应完成后,将化合物1的混合物通过微量过滤器逐滴添加至0.5%盐酸溶液中,该溶液也在20±5℃通过微米过滤器(30.0体积)缓慢过滤。将混合物搅拌至少4小时并离心。滤饼用纯净水(5.0体积×2)洗涤。
浆化操作:将dmso(9.0体积)和0.5%盐酸通过微米过滤器(30.0体积)加入反应器中,并将滤饼加入反应器中,并将混合物在20±5℃搅拌至少4小时,然后离心。将滤饼用纯净水(5.0体积×2)洗涤。将滤饼取样用于hplc分析,化合物1的标准≥98.0%。如果化合物1<98.0%,则重复“浆化操作”。将纯净水(40.0体积)和滤饼加入反应器中,并将混合物在20±5℃搅拌至少4小时,然后离心。将滤饼用纯净水(5.0体积×2)洗涤。然后将滤饼在45±5℃真空干燥至少8小时,直至lod≤3.0%。如果溶剂残留物不符合标准,则通过浆化除去残余溶剂:将纯净水(40.0体积)和产物加入反应器中,并将混合物在20±5℃搅拌至少4小时,然后离心。将滤饼用纯净水(5.0体积×2)洗涤。将滤饼在45±5℃真空干燥至少8小时,直至lod≤3.0%。对滤饼取样以获得溶剂残余物。如果溶剂残留物不符合标准,则重复“通过浆化除去残余溶剂”的操作,直到溶剂残留物符合标准。取样物质进行hplc分析,化合物1的标准≥98.0%(2.02kg)。将产品包装在带有干燥剂的双ldpe袋中,在室温储存。
化合物1:1hnmr(400mhz,dmso-d6)δ10.47(s,1h),8.54(s,1h),8.23–8.07(m,1h),7.96(d,j=5.6hz,1h),7.65–7.51(m,1h),7.23(s,1h),7.01(d,j=2.0hz,1h),6.96–6.87(m,2h),6.25(d,j=5.6hz,1h),4.98(d,j=6.0hz,1h),2.97(dd,j=5.6,1.6hz,1h),2.93(t,j=7.6hz,2h),2.54(t,j=7.6hz,2h),2.26(s,1h).
通过x射线粉末衍射图技术评价实施例1中制备的所得粉末的无定形或结晶性质。如图1中xrpd曲线中的结晶峰所证明,实施例1中制备的所得粉末被确定为结晶(在整个申请中有时称为“形式a”)。所得粉末也通过1h-nmr谱和13c-nmr谱表征,分别如图8和图9所示。
化合物1(形式a)的结晶形式的x射线粉末衍射图具有以下特征峰值衍射角(其中“间距”在图1中显示为“d值”):
表1:化合物1(形式a)的结晶形式的x射线粉末衍射图
形式a的长期稳定性
形式a的长期稳定性研究表明,在25℃/60%rh储存长达12个月(总杂质:t0=1.0%,t12=1.0%)和在40℃/75%rh条件下储存长达6个月(总杂质:t0=1.0%,t12=1.0%)时,没有发生明显的化学纯度变化。此外,当在25℃/60%rh储存长达12个月和在40℃/75%rh条件下储存长达6个月时,未观察到光学纯度变化。测试样品的xrpd数据显示,形式a在40℃/75%rh条件下在6个月时是稳定的,并且形式a在25℃/60%rh条件下在6个月时也是稳定的,但是在12个月时变为结晶形式(有时被称为“形式a*”)。
形式a*的xrpd如图2所示。化合物1的另一种晶形(形式a*)的x射线粉末衍射图具有以下特征峰衍射角(其中“间距”在图2中显示为“d值”):
表2:化合物1的另一种结晶形式(形式a*)的x射线粉末衍射图
稳定性研究表明,形式a是化学稳定的并且可以储存超过12个月而没有明显的分解。
如图10所示,还通过动态蒸汽吸附(dvs)评估形式a的吸湿性。图10显示形式a是适度吸湿的,在80%rh重量增量为4.96%。
正如下面的临床前研究中所讨论的,形式a在大鼠中的口服吸收相对较差,生物利用度为21%,部分原因是形式a溶解度低(发现在水中小于0.1μg/ml)。
由于在临床前研究中观察到的低生物利用度,因此形式a在直接药物制剂中的用途有限。然而,形式a是用于纯化api的良好候选物,并且用作制备无定形固体分散体的起始物质。尽管在上述条件下12个月时观察到结晶形式的变化(即从形式a到形式a*),但长期储存(即12个月)对后来的无定形mbp制备没有影响,因为化合物1无论如何在共沉淀发生之前溶解于dma中。
实施例1中制备的形式a的物理化学性质总结于表3中。
表3:形式a的主要物理化学性质
实施例2
化合物1(形式a**)的单晶
用于单晶x-射线衍射表征的化合物1etoac溶剂化物的板状单晶通过缓慢蒸发从etoac溶剂中结晶。实验细节详述如下。首先,将1.8mg化合物1称入3ml玻璃小瓶中,添加0.5mletoac溶剂。在涡旋振荡并超声摇动以加速溶解后,将悬浮液通过ptfe过滤膜(0.45μm)过滤,将滤液转移至干净的4ml壳瓶(44.6mmx14.65mm)中。随后,将壳瓶用其上有一个针孔的pe-plug密封,并放置在通风橱中,以在环境温度和湿度下缓慢蒸发。六天后,获得板状晶体样品。
使用从在etoac中缓慢冷却生长的单晶收集的一组衍射数据确定板状晶体的结构,并将其称为化合物1或形式a**的单晶。图3-6中列出了形式a**的晶体数据和结构精整。
表4:形式a**的单晶数据和结构精整
如图3所示,单一结构的不对称单元由两个独立的化合物1分子和两个etoac溶剂分子组成,表明晶体是化合物1的etoac溶剂化物。单晶结构测定证实,当以化合物1分子作为实例时,化合物1绝对构型为{c15(s),c16(s),c17(s)}。单晶的晶胞由四个化合物1分子和四个etoac溶剂分子组成,如图4所示。单晶结构中潜在的经典h键如图5所示。使用mercury软件计算的化合物1的单晶形式(即形式a**)的理论xrpd图的结果如图6所示。
实施例3
制备化合物1的纯无定形形式(形式b)
对化合物1的结晶形式a在dcm/meoh(2:1)中的溶液进行喷雾干燥,得到白色粉末。喷雾干燥的条件如下:将化合物1的结晶形式a(2.0g)在100ml混合溶剂(dcm/meoh=2:1体积比)中的溶液通过喷雾干燥器(buchi-290&buchi-295)进行喷雾干燥。产物粉末经红外灯在50℃干燥16小时。获得1.06g粉末。喷雾干燥器(buchi-290&buchi-295)的操作参数如下:入口温度:60℃;出口温度:35℃,抽气器:100%;泵%:15%;喷嘴清洁剂:2。
使用粉末x-射线衍射图方法表征所生成粉末的结构,证实其结构为无定形形式,例如图7的粉末x-射线衍射图无衍射峰。在本申请说明书中,化合物1的无定形形式称为化合物1的纯无定形形式或形式b。形式b的1h-nmr谱示于图11中。形式b的玻璃态转变温度测定为138.3℃。形式b的样品为白色粉末,其粒度为d90=69.9μm、d50=3.5μm、d10=1.4μm。测试样品的xrpd数据表明,形式b在40℃/75%rh的条件下经历14天是稳定的。例如,图12的xrpd数据证实了,该测试样品在14天时依然没有显示任何衍射峰。
实施例14
形式a、b在大鼠中药物动动力学对比。
1.药物和试剂:
微粉化后粒度为d90=62.4μm的形式a的粉末。物质含量(纯度)不低于98.0%。
微粉化后粒度为d90=69.9μm、d10=3.5μm、d50=1.4μm的形式b的粉末。物质含量(纯度)不低于98.0%。
2.实验动物:
本研究中使用雄性和雌性大鼠。
3.药物制备:
将适量的每种物质称重并分散在0.5%羧甲基纤维素钠中。对于每种物质,以期望浓度的化合物1制备悬浮液。在该研究中用游离碱计算化合物1的所有剂量和浓度。
4.给药和样品收集:
在剂量给药之前新鲜制备给药溶液。相应地记录实际体重和注射的实际体积。大鼠禁食过夜并在给药后4小时允许其摄取食物。将每种悬浮液以0.5至5mg/kg的剂量口服给予大鼠。在给药前和给药后直至36小时的不同时间通过头静脉丛收集血样(~1.0ml)。通过离心处理全血,收集血浆样品并在分析前保存在冰箱中。通过蛋白质沉淀处理血浆样品。使用经验证的液相色谱-串联质谱(lc-ms/ms)方法测定血浆样品中化合物1的浓度。使用pharsightwinnonlin使用非隔室模型分析血浆浓度-时间数据。每种物质的cmax和浓度-时间曲线下的面积示于表7中。
表7:大鼠中化合物1的形式a和b的口服给药pk曲线与形式a的静脉给药pk曲线
与结晶形式(形式a)相比,化合物1的纯无定形形式(形式b)也表现出更高的cmax(ng/ml)、auc0-inf(ng·h/ml)和f(%)。化合物1的纯无定形形式(形式b)的口服生物利用度达到了静脉注射的将近约50%,而结晶形式a的口服生物利用度只占静脉注射的约20%。这表明,本发明的纯无定形形式相对于结晶形式极大地提高了口服生物利用度。
以上实施例和实施方案的描述是示例性的,而非限制由权利要求书所限定的本发明。在不脱离如权利要求中所阐述的本发明的情况时,可以使用上述特征的多种变化和组合。所有这些变化都包括在本发明的范围内。引用的所有参考文献都通过引用整体并入本申请。
- 还没有人留言评论。精彩留言会获得点赞!