一种吲哚啉C7位乙酰氧基化的方法与流程
本发明涉及化合物的制备
技术领域:
,具体是一种吲哚啉c7位乙酰氧基化的方法。
背景技术:
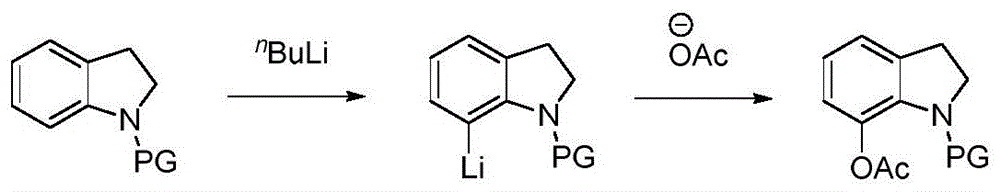
:吲哚啉,又称二氢吲哚,广泛存在于天然产物和药物分子中,是很多具有药理活性的生物碱的基本骨架。因此,对吲哚啉骨架的结构修饰及衍生化一直备受关注。传统的方法往往需要先将n原子用保护基团保护,然后使用强碱正丁基锂处理去氢得到c7位锂代的吲哚啉。最后亲核试剂处理可取代锂得到c7位官能团化的吲哚啉分子。图1所示为吲哚啉c7位乙酰氧基化的传统方法的反应路线示意图。该传统方法的主要缺点是需要使用强碱及易燃爆炸的极危险试剂正丁基锂,极大的限制了其应用。现有技术中另一种吲哚啉c7位乙酰氧基化的方法,即导向基团导向吲哚啉c7位c—h官能团化的策略,由于其高原子和步骤经济性,被广泛的使用。其中吲哚啉c7位的c—h乙酰氧基化可通过三价铑和二价银共催化实现。该方法的反应路线示意图如图2所示。尽管该方法避免了正丁基锂的使用,但是仍具有很大的缺陷:1)催化剂价格昂贵,需要使用两种催化剂,其中,(1,5-环辛二烯)氯化铑(i)二聚体(即百灵威试剂)的价格为1968元/g,氟化银的价格为692元/5g;2)需要使用的过量的强氧化剂(二乙酰氧基碘)苯的价格为855元/100g;3)以强腐蚀性乙酸酐(ac2o)作为反应溶剂。以制备100g7-乙酰氧基-n-嘧啶吲哚啉产物为例,该方法所需的催化剂和氧化剂成本大约为22350元。技术实现要素:本发明所要解决的技术问题是,针对现有技术的不足,提供一种经济、绿色的吲哚啉c7位乙酰氧基化的方法,该方法使用廉价的醋酸铜作为催化剂和乙酰氧基源,并以空气作为氧化剂,在特定的反应条件下在有机溶剂中进行c7位c—h官能团化,实现吲哚啉c7位c—h乙酰氧基化,得到c7位乙酰氧基化吲哚啉衍生物。本发明方法反应操作简单,反应成本低,产率高。本发明解决上述技术问题所采用的技术方案为:一种吲哚啉c7位乙酰氧基化的方法,以n-嘧啶基取代的二氢吲哚为原料,以空气作为氧化剂,以醋酸铜作为催化剂和乙酰氧基源,在有机溶剂中实现c7位c—h官能团化,得到c7位乙酰氧基化的吲哚啉产物。进一步地,本发明吲哚啉c7位乙酰氧基化的方法的具体过程为:将n-嘧啶基取代的二氢吲哚、醋酸铜和有机溶剂混合后,在装有空气的密闭容器中以120~140℃的反应温度反应12~48h,反应液冷却后,经过过滤、除去有机溶剂和柱层析纯化后得到c7位乙酰氧基化的吲哚啉产物。作为优选,所述的n-嘧啶基取代的二氢吲哚和醋酸铜的摩尔比为1:0.5~1。作为优选,所述的有机溶剂为乙腈(mecn)、四氢呋喃(thf)、1,4-二氧六环(1,4-dioxane)、n,n-二甲基甲酰胺(dmf)、二甲基亚砜(dmso)和二氯乙烷(dce)中的一种。进一步地,以所述的n-嘧啶基取代的二氢吲哚的用量为1当量,所述的醋酸铜的用量为0.5当量,所述的有机溶剂为二氯乙烷,所述的反应温度为130℃,反应时间为12h。作为优选,反应液冷却后加入乙酸乙酯,使用硅藻土过滤反应液,收集滤液,使用旋转蒸发仪除去滤液中的溶剂,最后使用石油醚与乙酸乙酯的体积比为3:1的柱色谱纯化后得到c7位乙酰氧基化的吲哚啉产物。与现有技术相比,本发明具有如下优点:本发明吲哚啉c7位乙酰氧基化的方法,以n-嘧啶基取代的二氢吲哚为原料,以空气作为氧化剂,以廉价的醋酸铜作为催化剂和乙酰氧基源,在特定的反应条件下在有机溶剂中进行c7位c—h官能团化,实现吲哚啉c7位c—h乙酰氧基化,得到c7位乙酰氧基化吲哚啉衍生物。本发明方法反应操作简单,反应成本低,产率高,当采用特定的反应条件组合时,产率最高可达95%。以一水合醋酸铜(价格为144元/100g)作为催化剂制备100g7-乙酰氧基-n-嘧啶吲哚啉产物为例,本发明方法所需的催化剂成本大约仅为60元,远低于以(1,5-环辛二烯)氯化铑(i)二聚体和氟化银作为催化剂的现有方法所需的22350元。附图说明图1为吲哚啉c7位乙酰氧基化的传统方法的反应路线示意图;图2为现有技术中另一种吲哚啉c7位乙酰氧基化的方法的反应路线示意图;图3为本发明吲哚啉c7位乙酰氧基化的方法的反应路线示意图;图4为实施例1的反应路线示意图;图5为实施例2的反应路线示意图;图6为实施例3的反应路线示意图;图7为实施例4的反应路线示意图;图8为实施例5的反应路线示意图;图9为实施例6的反应路线示意图;图10为实施例7的反应路线示意图;图11为实施例8的反应路线示意图。具体实施方式以下结合附图实施例对本发明作进一步详细描述。本发明吲哚啉c7位乙酰氧基化的方法,以n-嘧啶基取代的二氢吲哚为原料,以空气作为氧化剂,以醋酸铜作为催化剂和乙酰氧基源,在有机溶剂中实现c7位c—h官能团化,得到c7位乙酰氧基化的吲哚啉产物。本发明吲哚啉c7位乙酰氧基化的方法的反应路线示意图见图3。以n-嘧啶吲哚啉为例,为确定最佳反应条件,首先以n-嘧啶吲哚啉为模板底物,以n-嘧啶吲哚啉的用量为1当量,对反应条件进行优化,醋酸铜的用量、反应所用有机溶剂、反应温度和反应产率如表1所示。表1编号cu(oac)2的当量有机溶剂温度t(℃)产率(%)a11.0mecn1202721.0mecn1306031.0mecn1403841.0thf130551.01,4-dioxane1303561.0dmf1301071.0dmso130081.0dce1309190.5dce1309010b0.5dce13036a产率为分离纯化合物产率;b反应在氮气保护下进行。通过对醋酸铜的用量、反应所用有机溶剂、反应温度等条件的筛选,得到最佳的反应条件为0.5当量的cu(oac)2,在dce中加热130℃反应12h,n-嘧啶吲哚啉可实现c7位c—h官能团化,反应液冷却后加入乙酸乙酯,使用硅藻土过滤反应液,收集滤液,使用旋转蒸发仪除去滤液中的溶剂,最后使用石油醚与乙酸乙酯的体积比为3:1的柱色谱纯化后得到乙酰氧基化产物,产率为90%。实施例1,以确定的n-嘧啶吲哚啉模板底物的最佳反应条件为反应条件。取n-嘧啶吲哚啉1a(0.5mmol,98.6mg)、cu(oac)2·h2o(0.25mmol,50.0mg)溶解于dce(5.0ml),在装有空气的密闭容器中加热至130℃反应12h,反应液冷却后加入乙酸乙酯,使用硅藻土过滤反应液,收集滤液,使用旋转蒸发仪除去滤液中的溶剂,最后使用石油醚与乙酸乙酯的体积比为3:1的柱色谱纯化后得到白色固体产物2a,产率为90%。得到的产物的核磁参数为:1hnmr(400mhz,cdcl3)δ8.40(d,j=4.7hz,2h),7.10(d,j=6.6hz,1h),7.04-6.98(m,2h),6.70(t,j=4.7hz,1h),4.39(t,j=8.0hz,2h),3.13(t,j=8.0hz,2h),2.10(s,3h)ppm;13cnmr(101mhz,cdcl3)δ168.8,160.0,157.4,139.1,136.7,134.9,123.8,122.7,122.0,112.2,52.2,29.2,21.3ppm.实施例1的反应路线示意图见图4。实施例2,取5-甲氧基-n-嘧啶吲哚啉1b(0.5mmol,113.6mg)、cu(oac)2·h2o(0.25mmol,50.0mg)溶解于dce(5.0ml),在装有空气的密闭容器中加热至130℃反应12h,反应液冷却后加入乙酸乙酯,使用硅藻土过滤反应液,收集滤液,使用旋转蒸发仪除去滤液中的溶剂,最后使用石油醚与乙酸乙酯的体积比为3:1的柱色谱纯化后得到白色固体产物2b,产率为92%。得到的产物的核磁参数为:1hnmr(400mhz,cdcl3)δ8.37(d,j=4.6hz,2h),6.72(s,1h),6.65(t,j=4.6hz,1h),6.55(s,1h),4.38(t,j=7.8hz,2h),3.77(s,3h),3.07(t,j=7.7hz,2h),2.10(s,3h)ppm;13cnmr(101mhz,cdcl3)δ168.7,160.2,157.5,156.6,139.4,137.7,128.5,111.8,109.2,107.5,55.9,52.2,29.9,21.4ppm.hrmsm/z(esi)calcdforc15h15n3o3([m+h]+)285.1113,found285.1171.实施例2的反应路线示意图见图5。实施例3,取5-甲基-n-嘧啶吲哚啉1c(0.5mmol,105.6mg)、cu(oac)2·h2o(0.25mmol,50.0mg)溶解于dce(5.0ml),在装有空气的密闭容器中加热至130℃反应12h,反应液冷却后加入乙酸乙酯,使用硅藻土过滤反应液,收集滤液,使用旋转蒸发仪除去滤液中的溶剂,最后使用石油醚与乙酸乙酯的体积比为3:1的柱色谱纯化后得到白色固体产物2c,产率为95%。得到的产物的核磁参数为:1hnmr(400mhz,cdcl3)δ8.39(d,j=4.8hz,2h),6.93(s,1h),6.81(s,1h),6.67(t,j=4.8hz,1h),4.38(t,j=8.0hz,2h),3.08(t,j=8.0hz,2h),2.32(s,3h),2.10(s,3h)ppm;13cnmr(101mhz,cdcl3)δ168.9,160.1,157.5,138.8,136.7,134.0,132.5,123.0,122.9,112.0,52.3,29.3,21.4,20.8ppm.实施例3的反应路线示意图见图6。实施例4,取5-溴-n-嘧啶吲哚啉1d(0.5mmol,138.1mg)、cu(oac)2·h2o(0.25mmol,50.0mg)溶解于dce(5.0ml),在装有空气的密闭容器中加热至130℃反应24h,反应液冷却后加入乙酸乙酯,使用硅藻土过滤反应液,收集滤液,使用旋转蒸发仪除去滤液中的溶剂,最后使用石油醚与乙酸乙酯的体积比为3:1的柱色谱纯化后得到白色固体产物2d,产率为80%。得到的产物的核磁参数为:1hnmr(400mhz,dmso-d6)δ8.48(d,j=4.8hz,2h),7.36(s,1h),7.19(s,1h),6.92(t,j=4.8hz,1h),4.28(t,j=8.1hz,2h),3.13(t,j=8.0hz,2h),2.03(s,3h)ppm;13cnmr(101mhz,dmso-d6)δ168.1,159.5,158.2,139.5,139.2,135.1,125.8,125.2,113.7,113.6,52.5,28.8,21.4ppm.hrmsm/z(esi)calcdforc14h12brn3o2([m+h]+)334.0113,336.0113,found334.0172,336.0150.实施例4的反应路线示意图见图7。实施例5,取5-氟-n-嘧啶吲哚啉1e(0.5mmol,107.6mg)、cu(oac)2·h2o(0.25mmol,50.0mg)溶解于dce(5.0ml),在装有空气的密闭容器中加热至130℃反应12h,反应液冷却后加入乙酸乙酯,使用硅藻土过滤反应液,收集滤液,使用旋转蒸发仪除去滤液中的溶剂,最后使用石油醚与乙酸乙酯的体积比为3:1的柱色谱纯化后得到白色固体产物2e,产率为85%。得到的产物的核磁参数为:1hnmr(400mhz,cdcl3)δ8.45(d,j=4.8hz,2h),6.98(dd,j=9.0,4.8hz,1h),6.79-6.75(m,2h),4.46(t,j=8.1hz,2h),3.17(t,j=8.1hz,2h),2.11(s,3h)ppm;13cnmr(101mhz,cdcl3)δ168.4,158.7(d,j=221.9hz),157.5,155.2,136.9(d,j=6.9hz),134.9,124.3(d,j=8.7hz),112.8,122.0(d,j=23.5hz),110.5(d,j=22.6hz),52.4,29.6,21.3ppm.实施例5的反应路线示意图见图8。实施例6,取5-硝基-n-嘧啶吲哚啉1f(0.5mmol,127.1mg)、cu(oac)2·h2o(0.50mmol,100.0mg)溶解于dce(5.0ml),在装有空气的密闭容器中加热至130℃反应48h,反应液冷却后加入乙酸乙酯,使用硅藻土过滤反应液,收集滤液,使用旋转蒸发仪除去滤液中的溶剂,最后使用石油醚与乙酸乙酯的体积比为3:1的柱色谱纯化后得到黄色固体产物2f,产率为70%。得到的产物的核磁参数为:1hnmr(400mhz,cdcl3)δ8.49(d,j=4.7hz,2h),7.97(d,j=8.5hz,2h),6.87(t,j=4.7hz,1h),4.47(t,j=8.3hz,2h),3.24(t,j=8.2hz,2h),2.11(s,3h)ppm;13cnmr(101mhz,cdcl3)δ168.0,159.1,157.6,157.0,142.9,141.2,137.3,137.0,120.5,117.6,113.9,52.8,28.4,21.1ppm.实施例6的反应路线示意图见图9。实施例7,取3-甲基-n-嘧啶吲哚啉1g(0.5mmol,105.6mg)、cu(oac)2·h2o(0.25mmol,50.0mg)溶解于dce(5.0ml),在装有空气的密闭容器中加热至130℃反应12h,反应液冷却后加入乙酸乙酯,使用硅藻土过滤反应液,收集滤液,使用旋转蒸发仪除去滤液中的溶剂,最后使用石油醚与乙酸乙酯的体积比为3:1的柱色谱纯化后得到白色固体产物2g,产率为90%。得到的产物的核磁参数为:1hnmr(400mhz,cdcl3)δ8.39(d,j=4.7hz,2h),7.06–6.99(m,3h),6.69(t,j=4.7hz,1h),4.61-4.56(m,1h),3.91–3.86(m,1h),3.50–3.43(m,1h),2.09(s,3h),1.32(d,j=6.8hz,3h);13cnmr(101mhz,cdcl3)δ168.8,160.1,157.5,141.9,139.0,134.6,123.9,122.9,120.8,112.2,60.0,35.9,21.4,18.8ppm.实施例7的反应路线示意图见图10。实施例8,取2-甲基-n-嘧啶吲哚啉1h(0.5mmol,105.6mg)、cu(oac)2·h2o(0.25mmol,50.0mg)溶解于dce(5.0ml),在装有空气的密闭容器中加热至130℃反应12h,反应液冷却后加入乙酸乙酯,使用硅藻土过滤反应液,收集滤液,使用旋转蒸发仪除去滤液中的溶剂,最后使用石油醚与乙酸乙酯的体积比为3:1的柱色谱纯化后得到白色固体产物2h,产率为95%。得到的产物的核磁参数为:1hnmr(400mhz,dmso-d6)δ8.45(d,j=4.7hz,2h),7.16(d,j=7.1hz,1h),7.02(t,j=7.6hz,1h),6.95(d,j=8.0hz,1h),6.87(t,j=4.7hz,1h),4.90-4.83(m,1h),3.45(dd,j=15.8,8.8hz,1h),2.59(d,j=15.8hz,1h),2.01(s,3h),1.28(d,j=6.4hz,3h)ppm;13cnmr(101mhz,dmso-d6)δ168.1,159.4,158.2,139.7,135.8,133.8,123.7,123.0,122.6,113.3,59.7,36.4,21.5,21.1ppm.实施例8的反应路线示意图见图11。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1