化合物PaulownioneC和TomentodiplaconeO的合成方法与流程
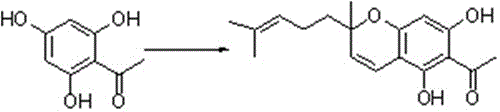
本发明涉及化合物合成及药物研发领域,具体涉及天然产物tomentodiplaconeo和paulownionec的合成方法。
背景技术:
tomentodiplaconeo和paulownionec是从paulowniatomentosa果实中分离出来的两个新的天然产物小分子,这两个天然产物分子属于带有苯并吡喃环结构的黄酮类化合物,因此具有黄酮的抗癌、抗肿瘤、杀菌、消炎等通性,进而使其具有了潜在的药用价值,而且对未来的药物开发或药物改进具有一定参考意义。然而,这两种天然产物的合成方法目前尚未见公开披露。
在苯并吡喃环类化合物的合成中,文献报道了用2,4,6-三羟基苯乙酮一水化合物与柠檬醛环化制备,但是这种反应操作中,需要使用毒性较高的干燥吡啶溶剂与催化剂。并且,反应过程中需要加热回流,反应过程中副产物种类较多,造成苯并吡喃环类化合物产物收率和选择性较低。
技术实现要素:
本发明的目的在于提供tomentodiplaconeo和paulownionec的合成方法,实现了对tomentodiplaconeo和paulownionec的首次绿色全合成。
本发明还在于解决了制备苯并吡喃化合物高毒低产的问题。
本发明采用相对低毒的甲醇做溶剂,避免使用高毒性的吡啶,廉价的无机碱如naoh做催化剂,在室温条件下制备出苯并吡喃底物,且在该合成过程中,反应产生的副产物仅有一种副产物,后续的分离简便,且产率较之前明显提高。
本发明中关键点主要包括(1)合成化合物6-乙酰基-5,7-二羟基-2-甲基-(4’-甲基-3’-戊烯基)-二氢-1-苯并吡喃;(2)6-乙酰基-5,7-二羟基-2-甲基-(4’-甲基-3’-戊烯基)-二氢-1-苯并吡喃、3,5-二甲氧基-4-羟基苯甲醛、3-甲氧基-4-羟基苯甲醛中羟基的保护;(3)合成含保护基的苯并吡喃环查尔酮化合物;(4)合成含保护基的苯并吡喃环黄烷酮化合物。
为实现上述目的,本发明采取的技术方案包括如下步骤:
(1)合成6-乙酰基-5,7-二羟基-2-甲基-(4’-甲基-3’-戊烯基)-二氢-1-苯并吡喃;将2,4,6-三羟基苯乙酮一水化合物加入至单口烧瓶,加入甲醇搅拌至溶质完全溶解,加入97%柠檬醛,再将naoh溶解在10ml甲醇后冰水浴滴加加入,1h后升至室温,搅拌反应24h,滴加3mol/l的盐酸溶液淬灭反应,调至溶液ph=3~4,用乙酸乙酯萃取,合并有机相后处理,柱层析分离得到6-乙酰基-5,7-二羟基-2-甲基-(4’-甲基-3’-戊烯基)-二氢-1-苯并吡喃;
(2)6-乙酰基-5,7-二羟基-2-甲基-(4’-甲基-3’-戊烯基)-二氢-1-苯并吡喃、3,5-二甲氧基-4-羟基苯甲醛、3-甲氧基-4-羟基苯甲醛中羟基的保护;将2,6-二羟基苯并吡喃环化合物、丁香醛、香草醛分别加入至单口圆底烧瓶,加入丙酮将底物溶解,再加入无水k2co3,回流半小时后冷却至室温,搅拌溶液并逐滴滴加氯甲基甲醚momcl,滴加完成后回流3h,用乙酸乙酯萃取,合并有机相后处理,柱层析分离得到三组带有甲氧基甲基mom保护基的底物,分别记为化合物三、化合物五、化合物十;
(3)含保护基的苯并吡喃环查尔酮化合物的合成;
将步骤(2)分离出的化合物三、化合物五、化合物十加入至单口圆底烧瓶,加入乙醇作为溶剂,在冰水浴n2氛围下,剧烈搅拌并缓慢加入koh-h2o-etoh溶液,滴加完成后搅拌1h后撤去冰水浴,室温条件下反应24h,用乙酸乙酯萃取,合并有机相后处理,柱层析分离得到两组含保护基的苯并吡喃环查尔酮化合物,分别记为化合物六、化合物十;
(4)含保护基的苯并吡喃环黄烷酮化合物的合成;
将步骤(3)分离出的化合物六、化合物十,加入到圆底单口烧瓶,加入无水etoh溶解,再加入naoac和一滴水,回流搅拌反应26h后,向反应体系中加入少量水,乙酸乙酯萃取,合并有机相用后处理,柱层析分离得到两组含保护基的苯并吡喃环黄烷酮化合物,分别记为化合物七、化合物十二;
(5)化合物paulownionec和tomentodiplaconeo的合成;
将步骤(4)合成的化合物七、化合物十二溶于5ml甲醇中,待完全溶解后加入3mol/l的盐酸1ml,加热回流,用薄层色谱监控反应,15min后撤去热源冷却至室温加水3ml,乙酸乙酯萃取,合并有机后处理,柱层析分离得到步骤(5)目标化合物。
步骤(1)中柠檬醛是2,4,6-三羟基苯乙酮一水化合物的1.2倍。
步骤(2)中k2co3的量是6-乙酰基-5,7-二羟基-2-甲基-(4’-甲基-3’-戊烯基)-二氢-1-苯并吡喃、3,5-二甲氧基-4-羟基苯甲醛、3-甲氧基-4-羟基苯甲醛总摩尔量的5倍,氯甲基甲醚momcl是6-乙酰基-5,7-二羟基-2-甲基-(4’-甲基-3’-戊烯基)-二氢-1-苯并吡喃、3,5-二甲氧基-4-羟基苯甲醛、3-甲氧基-4-羟基苯甲醛总摩尔量的1.2倍。
为了更好的解释本发明方案,结合附图,说明化合物三、化合物六、化合物七、化合物十、化合物十二等,并用(1)~(13)进行进一步的解释。
具体步骤可以为:
s1、6-乙酰基-5,7-二羟基-2-甲基-(4’-甲基-3’-戊烯基)-二氢-1-苯并吡喃(2)的合成。
将2,4,6-三羟基苯乙酮一水化合物1(3.7232g,20mmol)加入至250ml的单口烧瓶,加入甲醇(80ml)搅拌至溶质完全溶解,加入97%柠檬醛(3.7665g,24mmol),再将koh(3.3600g,60mmol)溶解在10ml甲醇后冰水浴滴加加入,1h后升至室温,搅拌反应24h,薄层色谱板监控反应,滴加3mol/l的盐酸溶液淬灭反应,调至溶液ph=3~4,用乙酸乙酯萃取(3×40ml),合并有机相用水洗(2×20ml),饱和食盐水洗(1×20ml),再将有机相用无水na2so4干燥之后减压蒸馏除去溶剂,柱层析分离得到目标化合物。
、6-乙酰基-5,7-二羟基-2-甲基-(4’-甲基-3’-戊烯基)-二氢-1-苯并吡喃(2)/3,5-二甲氧基-4-羟基苯甲醛(4)/3-甲氧基-4-羟基苯甲醛(9)中羟基的保护。
将2,6-二羟基苯并吡喃环化合物2/丁香醛4/香草醛9(10mmol)加入至50ml单口圆底烧瓶,加入丙酮(25ml)将底物溶解,再加入无水k2co3(1.6585g,12mmol),回流半小时后冷却至室温,搅拌溶液并逐滴滴加momcl(0.9660g,12mmol),滴加完成后回流3h,薄层色谱板监控反应,减压蒸馏除去溶剂,加少量水待固体全部溶解后转移分液漏斗,用乙酸乙酯萃取用乙酸乙酯萃取(3×30ml),合并有机相用水洗(2×20ml),饱和食盐水洗(1×20ml),再将有机相用无水na2so4干燥之后减压蒸馏除去溶剂,柱层析分离得到目标化合物。
、含保护基的苯并吡喃环查尔酮化合物6和11的合成。
将分别带有羟基保护基的苯并吡喃环底物3与丁香醛5/香草醛10加入至50ml单口圆底烧瓶,加入5ml乙醇作为溶剂,在冰水浴n2氛围下,剧烈搅拌并缓慢加入koh-h2o-etoh溶液,滴加完成后搅拌1h后撤去冰水浴,室温条件下反应24h,薄层色谱板监控反应,减压蒸馏除去溶剂,加少量水待固体全部溶解后转移分液漏斗,用乙酸乙酯萃取用乙酸乙酯萃取(3×30ml),合并有机相用水洗(2×20ml),饱和食盐水洗(1×20ml),再将有机相用无水na2so4干燥之后减压蒸馏除去溶剂,柱层析分离得到目标化合物。
、含保护基的苯并吡喃环黄烷酮化合物7和12的合成。
将带有保护基的苯并吡喃环查尔酮化合物6或11加入到25ml圆底单口烧瓶,加入无水etoh溶解,再加入naoac和一滴水,回流搅拌反应26h后,向反应体系中加入少量水,乙酸乙酯(3×30ml)萃取,合并有机相用水洗(2×20ml),饱和食盐水洗(1×20ml),再将有机相用无水na2so4干燥之后减压蒸馏除去溶剂,柱层析分离得到目标化合物。
、苯并吡喃环黄烷酮(化合物paulownionec(8)和tomentodiplaconeo(13))的合成。
将上步合成的含保护基的苯并吡喃环黄烷酮化合物7或12(0.5mmol)溶于5ml甲醇中,待完全溶解后加入3mol/l的盐酸1ml,加热回流,用薄层色谱监控反应,15min后撤去热源冷却至室温加水3ml,乙酸乙酯(3×30ml)萃取,合并有机相用水洗(2×20ml),饱和食盐水洗(1×20ml),再将有机相用无水na2so4干燥之后减压蒸馏除去溶剂,柱层析分离得到目标化合物。
附图说明
图1为本发明实施例中步骤s1的合成线路图。
图2为本发明实施例中步骤s2的合成线路图。
图3为本发明实施例中步骤s3的合成线路图。
图4为本发明实施例中步骤s4的合成线路图。
图5为本发明实施例中步骤s5的合成线路图。
图6为本发明实施例化合物paulownionec(8)的整体合成线路图。其中字母分别代表的化学操作及反应条件为:a)citral,naoh,meoh,rt;b)ch3och2cl,k2co3,acetone,reflux;c)koh-h2o-etoh,n2,rt;d)ch3coona,ch3ch2oh,reflux;e)hcl,meoh,50°c,rt,1h。
图7为本发明实施例化合物tomentodiplaconeo(13)的整体合成线路图。其中字母分别代表的化学操作及反应条件为:a)citral,naoh,meoh,rt;b)ch3och2cl,k2co3,acetone,reflux;c)koh-h2o-etoh,n2,rt;d)ch3coona,ch3ch2oh,reflux;e)hcl,meoh,50°c,rt,1h。
图8为发明实施例化合物paulownionec(8)的分子结构式及部分碳原子编号。
图9为发明实施例化合物tomentodiplaconeo(13)的分子结构式及部分碳原子编号。
具体实施方式
为了使本发明的目的及优点更加清楚明白,以下结合实施例对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
以下实施例中,nmr用bruker(400and100mhz)型核磁共振仪测定(cdcl3,cd3od,dmso作溶剂,tms为内标);硅胶为200-300目及gf254(青岛海洋化工厂生产);试剂均为分析纯。
如图1-5所示,本发明实施例提供了化合物paulownionec(8)和tomentodiplaconeo(13)的合成方法,包括如下步骤:
s1、6-乙酰基-5,7-二羟基-2-甲基-(4’-甲基-3’-戊烯基)-二氢-1-苯并吡喃(2)的合成
将2,4,6-三羟基苯乙酮一水化合物1(3.7232g,20mmol)加入至250ml的单口烧瓶,加入ch3oh(80ml)搅拌至溶质完全溶解,加入97%柠檬醛(3.7665g,24mmol),再将koh(3.3600g,60mmol)溶解在ch3oh(10ml)后冰水浴滴加加入,1h后升至室温,搅拌反应24h,薄层色谱板监控反应,滴加3mol/l的盐酸溶液淬灭反应,调至溶液ph=3~4,用乙酸乙酯萃取(3×40ml),合并有机相用水洗(2×20ml),饱和食盐水洗(1×20ml),再将有机相用无水na2so4干燥之后减压蒸馏除去溶剂,柱层析分离得到淡黄色油状化合物2(1.9932g,6.6mmol),产率33%。
、6-乙酰基-5,7-二羟基-2-甲基-(4’-甲基-3’-戊烯基)-二氢-1-苯并吡喃(2)/3,5-二甲氧基-4-羟基苯甲醛(4)/3-甲氧基-4-羟基苯甲醛(9)中羟基的保护。
将2,6-二羟基苯并吡喃环化合物2(2.9596g,9.8mmol)/丁香醛4(1.8218g,10mmol)/香草醛9(1.5215g,10mmol)加入至100ml单口圆底烧瓶,加入ch3coch3(40ml)将底物溶解,再加入无水k2co3(1.6585g,12mmol),回流半小时后冷却至室温,搅拌溶液并逐滴滴加momcl(0.9660g,12mmol),滴加完成后回流3h,薄层色谱板监控反应,减压蒸馏除去溶剂,加少量水待固体全部溶解后转移分液漏斗,用乙酸乙酯萃取(3×30ml),合并有机相用水洗(2×20ml),饱和食盐水洗(1×20ml),再将有机相用无水na2so4干燥之后减压蒸馏除去溶剂,柱层析分离得到淡黄色化合物3(2.577g,7.4mmol),产率76%/白色固体化合物5(1.8614g,8.2mmol)产率82%/白色固体化合物10(1.6464g,8.4mmol),产率84%。
、含保护基的苯并吡喃环查尔酮化合物(6)和(11)的合成。
将分别带有羟基保护基的苯并吡喃环底物3(0.6923g,2.0mmol)与化合物5(0.5445g,2.4mmol)/化合物10(0.4708,2.4mmol)加入至50ml单口圆底烧瓶,加入ch3ch2oh(20ml)作为溶剂,在冰水浴n2氛围下,剧烈搅拌并缓慢加入koh(5.6281g,100.5mmol)-h2o(5ml)-etoh(10ml)溶液,滴加完成后搅拌1h后撤去冰水浴,室温条件下反应24h,薄层色谱板监控反应,减压蒸馏除去溶剂,加少量水待固体全部溶解后转移分液漏斗,用乙酸乙酯萃取(3×30ml),合并有机相用水洗(2×20ml),饱和食盐水洗(1×20ml),再将有机相用无水na2so4干燥之后减压蒸馏除去溶剂,柱层析分离得到黄色油状化合物6(0.2386g,0.43mmol),产率为22%,黄色油状化合物11(0.1991g,0.38mmol),产率分为19%。
、含保护基的苯并吡喃环黄烷酮化合物(7)和(12)的合成。
将带有保护基的苯并吡喃环查尔酮化合物6(0.1128g,0.2mmol)化合物11(0.1048g,0.2mmol)加入到25ml圆底单口烧瓶,加入无水ch3ch2oh(5ml)溶解,再加入naoac(0.3286,4mmol)和一滴水,回流搅拌反应26h后,向反应体系中加入少量水,乙酸乙酯(3×30ml)萃取,合并有机相用水洗(2×20ml),饱和食盐水洗(1×20ml),再将有机相用无水na2so4干燥之后减压蒸馏除去溶剂,柱层析分离得到黄色油状化合物7(0.0577g,0.104mmol),产率为52%,黄色油状化合物12(0.0504g,0.096mmol),产率48%。
、苯并吡喃环黄烷酮(化合物paulownionec(8)和tomentodiplaconeo(13))的合成。
将上步合成的含保护基的苯并吡喃环黄烷酮化合物7(0.0376g,0.068mmol)或12(0.0397g,0.076mmol)溶于ch3oh(5ml)中,待完全溶解后加入3mol/l的盐酸(1ml),加热回流,用薄层色谱监控反应,15min后撤去热源冷却至室温加h2o(3ml),乙酸乙酯(3×30ml)萃取,合并有机相用水洗(2×20ml),饱和食盐水洗(1×20ml),再将有机相用无水na2so4干燥之后减压蒸馏除去溶剂,柱层析分离得到黄色油状化合物paulownionec(8)(0.0149g,0.032mmol),产率47%,黄色油状化合物tomentodiplaconeo(13)(0.0164g,0.037mmol),产率49%。
(8):1hnmr(cd3od,400mhz)δh:6.77(s,2h,h-2’,h-6’),6.59(d,j=10.0hz,1h,h-1’’),5.98(s,1h,h-8),5.47(d,j=10.0hz,1h,h-2’’),5.31(brd,j=12.4hz,1h,h-2),5.11(t,1h,j=7.0hz,h-7’’),3.86(s,6h,meo-3’,meo-5’),3.03(dd,1h,j=12.8,17.0hz,h-3β),2.65(brd,1h,j=17.0hz,h-3α),2.16(m,2h,h-6’’),1.75(m,2h,h-5’’),1.66(s,3h,h-9’’),1.50(s,3h,h-10’’),1.41(s,3h,h-4’’);13cnmr(cd3od,400mhz)δc:192.6(c-4),165.9(c-9),162.1(c-7),158.8(c-5),150.1(c-3’,c-5’),137.7(c-4’),133.3(c-8’’),132.1(c-1’),126.9(c-2’’),126.2(c-7’’),118.5(c-1’’),106.7(c-2’,c-6’),106.2(c-6),105.8(c-10),97.2(c-8),82.1(c-3’’),81.5(c-2),57.4(meo-3’,meo-5’),47.7(c-3),42.8(c-5’’),27.5(c-4’’),24.5(c-9’’),18.5(c-6’’),15.3(c-10’’)
tomentodiplaconeo(13):1hnmr(cd3od,400mhz)δh:7.06(brs,1h,h-2’),6.91(brd,j=8.0hz,h-6’),6.81(d,1h,j=8.0hz,h-5’),6.58(d,1h,j=10.0hz,h-1’’),5.97(s,1h,h-8),5.46(d,1h,j=10.0hz,h-2’’),5.31(brd,1h,j=12.5hz,h-2),5.12(t,1h,j=7.4hz,h-7’’),3.87(s,3h,meo-3’),3.02(dd,1h,j=12.5,17.0hz,h-3β),2.64(brd,1h,j=17.0hz,h-3α),2.16(q,2h,j=7.4hz,h-6’),1.64(m,2h,h-5’’),1.57(s,3h,h-9’’),1.56(s,3h,h-10’’),1.42(s,1h,h-4’’);13cnmr(cd3od,400mhz)δc:191.8(c-4),165.1(c-9),161.3(c-7),157.9(c-5),149.0(c-3’),148.0(c-4’),132.4(c-8’’),132.0(c-1’),126.0(c-2’’),125.4(c-7’’),120.4(c-6),117.6(c-1’’),116.1(c-5’’),111.1(c-2’),105.9(c-6),105.3(c-10),96.2(c-8),81.2(c-3’’),80.4(c-2),56.4(meo-3’),42.0(c-3),41.8(c-5’’),26.6(c-4’’),25.8(c-9’’),23.7(c-6’’),17.7(c-10’’).
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以作出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!