一种3-(噻吩-2-基)环己酮骨架化合物的制备方法与流程
本发明属于噻吩取代的环己酮衍生物制备方法技术领域,具体涉及一种3-(噻吩-2-基)环己酮骨架化合物的制备方法。
背景技术:
噻吩类化合物是天然产物、农产品、医药、多聚物以及精细化学品等的重要骨架[参见:j.med.chem.2016,59,8830.]。环己烯酮类化合物是天然产物、生物活性及药物活性分子中重要的结构单元[参见:j.nat.prod.2016,79,74-80.]。关于噻吩取代的环己酮类化合物的合成受到化学家的广泛关注。目前关于合成3-(噻吩-2-基)环己酮类化合物的方法包括:大位阻的环己烯酮与芳基硼化物的共轭加成[参见:j.org.lett.2012,14,2508.];环己烯酮和芳基硼酸的共轭加成[参见:org.lett.2013,15,1886.];环己烯酮与芳基硅试剂的共轭加成[参见:org.lett.2007,9,2737.];环己烯酮与噻吩被金属锌取代的化合物的共轭加成[参见:chem.commun.,2008,3795–3797.];环己酮和芳基碘的直接β-位官能团化等[参见:j.am.chem.soc.2013,135,17747.],但这些方法往往都需要使用预先官能团化的底物或者对空气、水敏感试剂,这严重限制了β-位环己烯酮的官能团化反应以及应用潜力。因此,急需开发一种经济、高效的策略,利用未预官能团化的底物来制备3-位杂环基环己酮衍生物。
技术实现要素:
本发明的目的是提供一种3-(噻吩-2-基)环己酮骨架化合物的制备方法,该合成方法具有原料廉价易得、反应步骤简单、原子利用率高等优点。
本发明为实现上述目的而采取的技术方案为:
一种3-(噻吩-2-基)环己酮骨架化合物的制备方法,包括如下步骤:
(1)按摩尔比1:2:0.1:0.2将噻吩或取代的噻吩、环己烯酮、醋酸钯、配体加入反应器中,加入溶剂将反应物溶解,在室温下混合均匀,在50℃下反应24~36小时;
(2)反应完成后将反应器冷却至室温,向反应器中加入乙酸乙酯稀释反应物,之后将反应物过滤并旋去溶剂;将旋去溶剂后的剩余物分离纯化,旋蒸除去溶剂,油泵抽干,得到目标产物3-(噻吩-2-基)环己酮骨架化合物。
所述取代的噻吩为2-溴噻吩、2-氯噻吩、2-甲基噻吩、2-苯基噻吩、4-溴-2-甲基噻吩、2-溴-3-甲基噻吩或2-氯-3-甲基噻吩。
步骤(1)中所述的溶剂为冰醋酸和乙腈的混合溶剂。
所述冰醋酸和乙腈的体积比为20:1。
步骤(1)中所述配体为4,5-二氮芴-9-酮。
步骤(2)中将采用乙酸乙酯稀释后的反应物通过玻璃砂芯漏斗过滤。
步骤(2)中将旋去溶剂后的剩余物采用硅胶柱层析分离纯化。
本发明的技术路线是取代的噻吩与环己烯酮的共轭加成反应,其化学方程式为:
其中,r为甲基、甲氧基、氯、溴等。
本发明采用核磁共振氢谱(1hnmr)、碳谱(13cnmr)以及高分辨质谱证实了3-(噻吩-2-基)环己酮骨架化合物的结构。检测所用仪器为:avanceiiihd600mhz型核磁共振仪,其中氘代氯仿为内标(氢谱,氘代氯仿:δ7.26ppm).(碳谱,氘代氯仿:δ77ppm)。thermoscientificqexactive型高分辨质谱仪。
与现有合成方法相比,本发明的优点具体体现为:
(1)本发明首次实现了钯催化下噻吩类化合物与环己烯酮的共轭加成反应,与传统合成方法相比较,该方法原料无需经过多步预官能化步骤来制备,反应步骤简单,合成效率高;
(2)本发明所述反应底物的官能团适用范围较广,包括:含卤素、烷基、烷氧基等官能团底物;
(3)本发明所用合成路线为快速高效得到含有3-(噻吩-2-基)环己酮骨架结构的复杂天然产物以及药物分子提供了新的途径。
附图说明
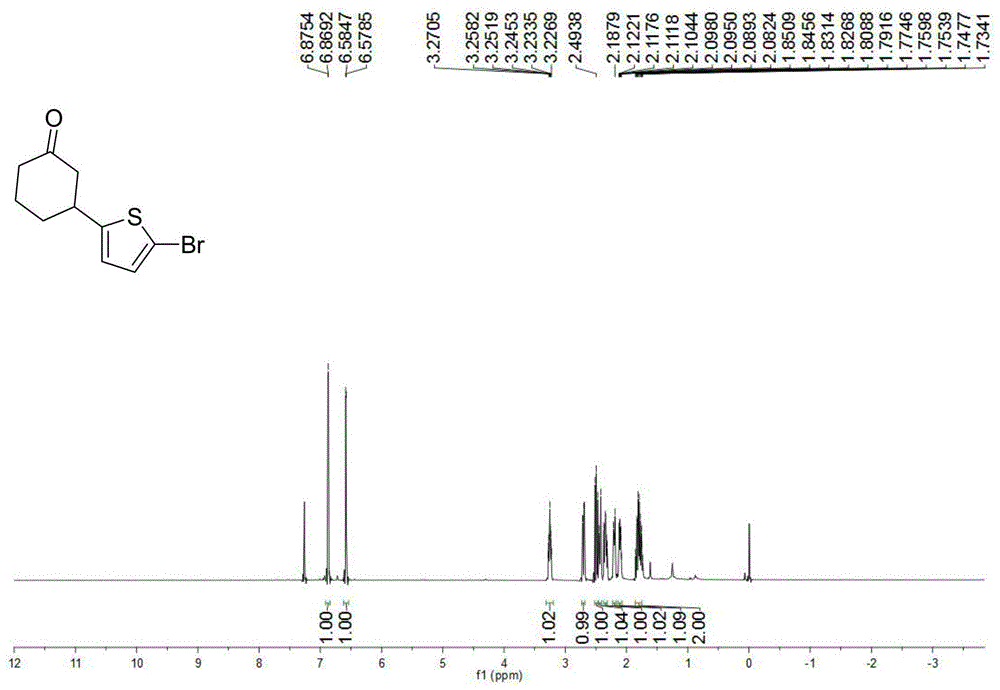
图1为3-(5-溴噻吩-2-基)环己酮的氢谱;
图2为3-(5-溴噻吩-2-基)环己酮的碳谱;
图3为3-(5-氯噻吩-2-基)环己酮的氢谱;
图4为3-(5-氯噻吩-2-基)环己酮的碳谱;
图5为3-(5-甲基噻吩-2-基)环己酮的氢谱;
图6为3-(5-甲基噻吩-2-基)环己酮的碳谱;
图7为3-(5-苯基噻吩-2-基)环己酮的氢谱;
图8为3-(5-苯基噻吩-2-基)环己酮的碳谱;
图9为3-(5-溴-4-甲基噻吩-2-基)环己酮的氢谱;
图10为3-(5-溴-4-甲基噻吩-2-基)环己酮的碳谱。
图11为3-(5-氯-4-甲基噻吩-2-基)环己酮的氢谱;
图12为3-(5-氯-4-甲基噻吩-2-基)环己酮的碳谱;
图13为3-(3-溴-5-甲基噻吩-2-基)环己酮的氢谱;
图14为3-(3-溴-5-甲基噻吩-2-基)环己酮的碳谱。
具体实施方式
下面结合具体实施例对本发明作进一步详细描述,将有助于对本发明的理解,但并不是以此来限制本发明的权利范围。
实施例1:3-(5-溴噻吩-2-基)环己酮的合成
(1)将2-溴噻吩(0.024ml,0.25mmol),环己烯酮(0.048ml,0.50mmol),醋酸钯(0.0056g,0.025mmol),4,5-二氮芴-9-酮(0.0090g,0.05mmol),冰醋酸(0.6ml),乙腈(0.03ml),在干净干燥的密闭反应管中搅拌均匀后加热到50℃,反应24小时。
(2)反应完成后将反应管冷却至室温,加入50ml的乙酸乙酯稀释并通过玻璃砂芯漏斗过滤,在旋转蒸发仪上旋去溶剂,将旋去溶剂后的剩余物用硅胶柱层析(石油醚/乙酸乙酯=20:1-10:1,v/v)分离纯化,旋蒸除去溶剂,油泵抽干,得到黄色油状物,目标产物38.6mg,产率60%。氢谱和碳谱如图1和图2所示,1hnmr(600mhz,cdcl3)δ6.87(d,j=3.7hz,1h),6.58(d,j=3.7hz,1h),3.28–3.23(m,1h),2.72–2.69(m,1h),2.52–2.47(m,1h),2.43(d,j=14.9hz,1h),2.37–2.31(m,1h),2.22–2.18(m,1h),2.12–2.08(m,1h),1.85–1.73(m,2h);13cnmr(151mhz,cdcl3)δ209.42,149.84,129.46,123.35,109.72,48.84,40.96,39.93,33.39,24.68.hrms(esi):计算值c10h11brosna[m+na]+280.9606,实测值280.9615。
实施例2:3-(5-氯噻吩-2-基)环己酮的合成。
(1)将2-氯噻吩(0.023ml,0.25mmol),环己烯酮(0.048ml,0.50mmol),醋酸钯(0.0056g,0.025mmol),4,5-二氮芴-9-酮(0.0090g,0.05mmol),冰醋酸(0.6ml),乙腈(0.03ml),在干净干燥的密闭反应管中搅拌均匀后加热到50℃,反应24小时。
(2)反应完成后将反应管冷却至室温,加入50ml的乙酸乙酯稀释并通过玻璃砂芯漏斗过滤,在旋转蒸发仪上旋去溶剂,将旋去溶剂后的剩余物用硅胶柱层析(石油醚/乙酸乙酯=20:1-10:1,v/v)分离纯化,旋蒸除去溶剂,油泵抽干,得到棕色油状物,目标产物33.6mg,产率63%。氢谱和碳谱如图3和图4所示,1hnmr(600mhz,cdcl3)δ6.73(d,j=3.7hz,1h),6.59(dd,j=3.7,0.9hz,1h),3.25–3.20(m,1h),2.72–2.68(m,1h),2.51–2.47(m,1h),2.45–2.41(m,1h),2.37–2.31(m,1h),2.21–2.18(m,1h),2.13–2.08(m,1h),1.84–1.74(m,2h);13cnmr(151mhz,cdcl3)δ209.45,146.93,127.63,125.66,122.28,48.83,40.97,39.92,33.40,24.68.hrms(esi):计算值c10h12clos[m+h]+215.0292,实测值215.0289。
实施例3:3-(5-甲基噻吩-2-基)环己酮的合成
(1)将2-甲基噻吩(0.024ml,0.25mmol),环己烯酮(0.048ml,0.50mmol),醋酸钯(0.0056g,0.025mmol),4,5-二氮芴-9-酮(0.0090g,0.05mmol),冰醋酸(0.6ml),乙腈(0.03ml),在干净干燥的密闭反应管中搅拌均匀后加热到50℃,反应24小时。
(2)反应完成后将反应管冷却至室温,加入50ml的乙酸乙酯稀释并通过玻璃砂芯漏斗过滤,在旋转蒸发仪上旋去溶剂,将旋去溶剂后的剩余物用硅胶柱层析(石油醚/乙酸乙酯=20:1-10:1,v/v)分离纯化,旋蒸除去溶剂,油泵抽干,得到黄色油状物,目标产物34.6mg,产率70%。氢谱和碳谱如图5和图6所示,1hnmr(600mhz,cdcl3)δ5.59(d,j=3.2hz,1h),5.57(d,1h),2.27–2.22(m,1h),1.71(dd,j=13.5,2.8hz,1h),1.51(t,j=12.9hz,1h),1.44(s,3h),1.41(s,1h),1.37–1.31(m,1h),1.21–1.19(m,1h),1.12–1.08(m,1h),0.86–0.72(m,2h);13cnmr(151mhz,cdcl3)δ210.18,145.99,137.71,124.60,122.53,49.28,41.06,39.83,33.67,24.83,15.26.hrms(esi):计算值c11h15os[m+h]+195.0838,实测值195.0838。
实施例4:3-(5-苯基噻吩-2-基)环己酮的合成
(1)将2-苯基噻吩(0.0400g,0.25mmol),环己烯酮(0.048ml,0.50mmol),醋酸钯(0.0056g,0.025mmol),4,5-二氮芴-9-酮(0.0090g,0.05mmol),冰醋酸(0.6ml),乙腈(0.03ml),在干净干燥的密闭反应管中搅拌均匀后加热到50℃,反应24小时。
(2)反应完成后将反应管冷却至室温,加入50ml的乙酸乙酯稀释并通过玻璃砂芯漏斗过滤,在旋转蒸发仪上旋去溶剂,将旋去溶剂后的剩余物用硅胶柱层析(石油醚/乙酸乙酯=20:1-10:1,v/v)分离纯化,旋蒸除去溶剂,油泵抽干,得到黄色油状物,目标产物44.1mg,产率69%。氢谱和碳谱如图7和图8所示,1hnmr(600mhz,cdcl3)δ7.55(dd,j=8.2,1.0hz,2h),7.35(t,j=7.7hz,2h),7.25(dd,j=8.7,6.2hz,1h),7.12(d,j=3.7hz,1h),6.78(dd,j=3.6,0.7hz,1h),3.34–3.29(m,1h),2.78–2.74(m,1h),2.58–2.54(m,1h),2.46–2.42(m,1h),2.38–2.33(m,1h),2.26–2.23(m,1h),2.15–2.10(m,1h),1.92–1.85(m,1h),1.82–1.75(m,1h);13cnmr(151mhz,cdcl3)δ209.85,147.81,142.22,134.31,128.81,127.29,125.55,123.87,122.60,49.10,41.04,39.90,33.57,24.80.hrms(esi):计算值c16h15o2[m+h]+257.0995,实测值257.0992。
实施例5:3-(5-溴-4-甲基噻吩-2-基)环己酮的合成
(1)将2-溴3-甲基噻吩(0.028ml,0.25mmol),环己烯酮(0.048ml,0.50mmol),醋酸钯(0.0056g,0.025mmol),4,5-二氮芴-9-酮(0.0090g,0.05mmol),冰醋酸(0.6ml),乙腈(0.03ml),在干净干燥的密闭反应管中搅拌均匀后加热到50℃,反应36小时。
(2)反应完成后将反应管冷却至室温,加入50ml的乙酸乙酯稀释并通过玻璃砂芯漏斗过滤,在旋转蒸发仪上旋去溶剂,将旋去溶剂后的剩余物用硅胶柱层析(石油醚/乙酸乙酯=20:1-10:1,v/v)分离纯化,旋蒸除去溶剂,油泵抽干,得到黄色油状物,目标产物49.2mg,产率72%。氢谱和碳谱如图9和图10所示,1hnmr(600mhz,cdcl3)δ6.51(s,1h),3.23–3.18(m,1h),2.70–2.66(m,1h),2.50–2.46(m,1h),2.44–2.41(m,1h),2.36–2.31(m,1h),2.19–2.16(m,1h),2.12(s,3h),2.11–2.09(m,1h),1.83–1.74(m,2h);13cnmr(151mhz,cdcl3)δ209.55,147.51,136.76,125.18,106.66,48.74,40.99,39.89,33.31,24.66,15.20.hrms(esi):计算值c11h14bros[m+h]+272.9943,实测值272.9940。
实施例6:3-(5-氯-4-甲基噻吩-2-基)环己酮的合成
(1)将2-氯-3-甲基噻吩(0.027ml,0.25mmol),环己烯酮(0.048ml,0.50mmol),醋酸钯(0.0056g,0.025mmol),4,5-二氮芴-9-酮(0.0090g,0.05mmol),冰醋酸(0.6ml),乙腈(0.03ml),在干净干燥的密闭反应管中搅拌均匀后加热到50℃,反应36小时。
(2)反应完成后将反应管冷却至室温,加入50ml的乙酸乙酯稀释并通过玻璃砂芯漏斗过滤,在旋转蒸发仪上旋去溶剂,将旋去溶剂后的剩余物用硅胶柱层析(石油醚/乙酸乙酯=20:1-10:1,v/v)分离纯化,旋蒸除去溶剂,油泵抽干,得到黄色油状物,目标产物44.9mg,产率78%。氢谱和碳谱如图11和图12所示,1hnmr(600mhz,cdcl3)δ6.49(s,1h),3.20–3.16(m,1h),2.68(dd,j=14.7,2.0hz,1h),2.48(d,j=12.7hz,1h),2.45–2.41(m,1h),2.36–2.31(m,1h),2.18–2.16(m,1h),2.12(s,3h),2.09(d,j=5.0hz,1h),1.82–1.73(m,2h);13cnmr(151mhz,cdcl3)δ209.57,144.40,133.82,124.77,122.29,48.74,40.99,39.81,33.33,24.65,13.55.hrms(esi):计算值c11h14clos[m+h]+229.0448,实测值229.0446。
实施例7:3-(3-溴-5-甲基噻吩-2-基)环己酮的合成
(1)将4-溴-2-甲基噻吩(0.028ml,0.25mmol),环己烯酮(0.048ml,0.50mmol),醋酸钯(0.0056g,0.025mmol),4,5-二氮芴-9-酮(0.0090g,0.05mmol),冰醋酸(0.6ml),乙腈(0.03ml),在干净干燥的密闭反应管中搅拌均匀后加热到50℃,反应36小时。
(2)反应完成后将反应管冷却至室温,加入50ml的乙酸乙酯稀释并通过玻璃砂芯漏斗过滤,在旋转蒸发仪上旋去溶剂,将旋去溶剂后的剩余物用硅胶柱层析(石油醚/乙酸乙酯=20:1-10:1,v/v)分离纯化,旋蒸除去溶剂,油泵抽干,得到黄色油状物,目标产物49.2mg,产率72%。氢谱和碳谱如图13和图14所示,1hnmr(600mhz,cdcl3)δ6.58(d,j=1.0hz,1h),3.40–3.36(m,1h),2.69–2.66(m,1h),2.47–2.44(m,1h),2.43(d,j=0.9hz,3h),2.40–2.37(m,1h),2.36–2.32(m,1h),2.16–2.13(m,2h),1.82–1.75(m,2h);13cnmr(151mhz,cdcl3)δ209.32,139.56,137.64,127.78,106.46,48.42,40.95,39.42,32.50,25.08,15.42.hrms(esi):计算值c10h11brclos[m+h]+292.9397,实测值292.9402。
- 还没有人留言评论。精彩留言会获得点赞!