一种耐酸型单宁酶及其基因和应用的制作方法
本发明属于生物工程技术领域,具体涉及一种耐酸型单宁酶及其基因和应用。
背景技术:
单宁酶的应用十分广泛,在食品工业中,不仅应用于饮料的澄清、防止其中酯类变质、还可以对一些饮料中的麦芽多糖进行稳定处理、风味的改进,以及作为测定天然没食子酸酯类结构的一种灵敏的分析探针;在饲料加工过程中如用单宁酶处理,可以大大提高家畜对植物蛋白的利用率,降低畜牧业的成本;没食子酸(ga)是单宁酶水解底物中产生的主要产物,是制药工业中的甲氧苄胺嘧啶(trimethoprim)化合物和食品中抗氧化剂没食子酸丙酯(pg)合成所必需的重要前体物质,近年来还可以用于半导体感光树脂的原料等。因此对单宁酶的有效利用和深层开发已成为目前国内外的研究热点。
20世纪20年代,freudenberg在进行黑曲霉培养时发现在其菌丝中含有单宁酶;yosuken等(2004)从人粪和发酵了的食物中分离出了产单宁酶的乳酸杆菌。bartaa等(2005)对35株曲霉和25株青霉的产单宁酶能力进行了分析研究,以筛选出新的产单宁酶品种。但从自然界中分离筛选得到的菌种产单宁酶的活力不是很高,因此采用现代生物技术构建出单宁酶高产工程菌是提高单宁酶活力的一个主要途径。hatameto等(1996)克隆出米曲霉的单宁酶基因,并将带有单宁酶基因的质粒转化至单宁酶产量低的菌株aspergillusorygzeaol,提高其单宁酶产量;这为构建重组系统高效表达单宁酶打下了基础。
现有的单宁酶的适应ph值在偏酸和中性范围内,最适温度在20-60℃之间。这使得单宁酶在食品生产,尤其是酸性果汁饮料处理的过程中受限制。因而现有的单宁酶生产技术仍有待改进。
技术实现要素:
本发明旨在至少在一定程度上解决上述技术中的技术问题之一。为此,本发明的第一个目的在于提出一种编码耐酸型单宁酶的基因。该耐酸型单宁酶具有在酸性条件下高活力的特点,符合在食品工业中尤其酸性果汁饮料处理的工业要求。
本发明的第二个目的是提出一种表达载体。
本发明的第三个目的是提出一种重组菌株。
本发明的第四个目的是提出一种制备耐酸型单宁酶的方法。
本发明的第五个目的是提出耐酸型单宁酶的用途。
在本发明的第一个方面中,根据本发明实施例,提供一种编码耐酸型单宁酶编码的基因,该耐酸型单宁酶的氨基酸序列如seqidno:1所示,所述基因的核酸序列如seqidno:2所示。
本发明基于热不对称pcr的方法分离克隆了该耐酸型单宁酶tan1的核酸序列,序列分析结果表明,耐酸型单宁酶结构基因全长1713bp,前51位为信号肽编码序列,无内含子序列。本发明从得到的单宁酶基因通过blast比对与前人公开的序列不同,是一个新序列。
本发明的耐酸型单宁酶tan1,其氨基酸序列如seqidno:1所示。其中,该酶包括570个氨基酸和一个终止密码子,n端分析前17位氨基酸为信号肽序列,去除信号肽后,成熟的耐酸型单宁酶的理论分子量为61.14kda。本发明获得的单宁酶具有生物活性,酶活力为49.75u/ml,以及具有较好的耐酸性。
根据本发明实施例的耐酸型单宁酶tan1,其最适ph值为5.0,在ph2~7的范围内稳定;最适温度为50℃,在30~50℃范围内具有较高的酶活力。相比于文献来源单宁酶tan2(tan2,yuxw,liyq,etal.2008),酶tan1具有在酸性条件下高活力的特点,符合在食品工业中尤其酸性果汁饮料处理的工业要求。除此之外,其宽泛的温度活性范围在饲料、医药和废水处理等领域具有潜在的应用价值。
在本发明的第二方面中,根据本发明实施例,提供了一种表达载体,其包含上述编码耐酸型单宁酶的多核苷酸。其中,多核苷酸为dna序列。
在本发明的第三方面中,根据本发明实施例,提供了一种重组菌株,由前述表达载体转化宿主细胞获得。其中,宿主细胞采用毕赤酵母gs115。
在本发明的第四方面中,根据本发明实施例,提供了一种制备耐酸型单宁酶的方法,其包括下列步骤:
(1)、以黑曲霉基因组dna为模板,通过设计两对特异性引物,利用热不对称pcr的方法扩增单宁酶基因序列;其中,黑曲霉的保藏编号为cctccno:m2019358,保藏名称为黑曲霉aspergillusnigerfj0118;
(2)、将所述单宁酶基因克隆到质粒中,得到上述表达载体;
(3)、将所述表达载体转化宿主细胞,得到上述重组菌株;
(4)、将所述重组菌株在发酵罐上诱导表达,得到所述耐酸型单宁酶。
根据本发明的进一步实施例,在步骤(1)中,所述特异性引物为:
上游特异性引物:qtan-f1的序列如seqidno:3所示,qtan-f2的序列如seqidno:4;
下游特异性引物:qtan-r1的序列如seqidno:5所示,qtan-r2的序列如seqidno:6。
根据本发明的进一步实施例,所述步骤(4)中诱导耐酸型单宁酶表达采用甲醇发酵罐进行诱导表达,所述甲醇发酵罐诱导表达包括:甘油分批发酵阶段、饥饿阶段及甲醇流加阶段。
根据本发明的进一步实施例,还包括分离纯化所述耐酸型单宁酶,所述分离纯化为:采用1.6×20cm的阴离子交换柱纯化蛋白,用柠檬酸缓冲液平衡柱子,流速为1ml/min;加入耐酸型单宁酶粗酶液,流速为1ml/min,孵育20min;用含nacl的柠檬酸缓冲液洗脱,流速为1ml/min,用收集并浓缩各步洗脱液,sds-page分析蛋白纯化情况。
根据本发明实施例,本发明通过设计两对特异引物,利用热不对称pcr的方法将编码黑曲霉单宁酶成熟蛋白的核酸序列从黑曲霉基因组中扩增出来,克隆到载体上,获得一定拷贝数后,酶切酶连至毕赤酵母表达载体ppic9k,构建重组表达载体ppic9k-tan1,采用电击法转化毕赤酵母gs115中,采用甲醇进行诱导表达,离心的上清中的单宁酶再进行纯化得到耐酸型单宁酶。
由此,表达载体ppic9k-tan1可以在甲醇的诱导条件下高效表达目的酶蛋白;毕赤酵母表达系统自身分泌的背景蛋白少,易于纯化,且采用deae阴离子交换层析一步纯化蛋白,减少酶蛋白损失;毕赤酵母表达系统糖基化程度低,对营养要求低,可采用廉价的培养基,可实现高密度发酵;利用本发明的方法获得的重组单宁酶可处理低ph的果汁样品,具有除涩澄清作用;且在不控ph条件下几乎完全降解单宁酸生产没食子酸,大大简化了工艺操作。
在本发明的第五方面中,根据本发明实施例,本发明还提出了上述耐酸型单宁酶在食品工业中的应用,将上述耐酸型单宁酶用在酸性果汁饮料的处理过程中,降低酯型儿茶素的含量,表明此耐酸型单宁酶可以应用于酸性果汁处理。
在本发明的第六方面中,根据本发明实施例,本发明还提出了上述耐酸型单宁酶在生产没食子酸中的应用。
本发明的附加方面和优点将在下面的描述中部分给出,部分将从下面的描述中变得明显,或通过本发明的实践了解到。
附图说明
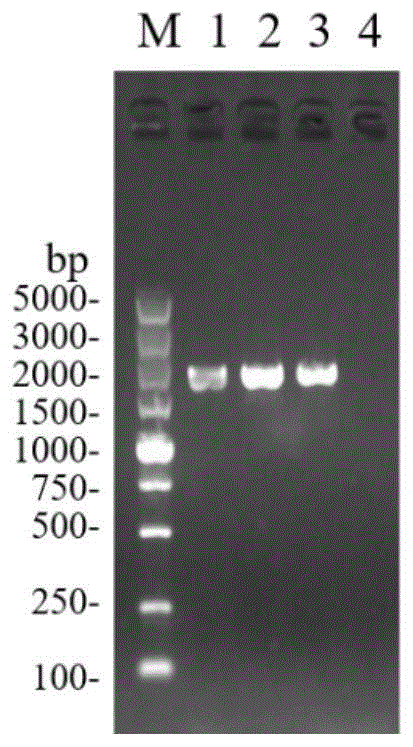
图1为黑曲霉单宁酶tan1基因pcr扩增电泳结果;m:dl5000marker,1-3:梯度扩增产物;
图2为菌液pcr鉴定pmd19t-tan1克隆载体;其中m:dl5000marker,1-8:通用性引物扩增结果,9-16:特异性引物扩增结果;
图3为线性化表达载体ppic9k-tan1电泳结果;m:dl15000marker,1:未线性化质粒,2:线性化质粒;
图4为破壁pcr鉴定阳性酵母转化子电泳结果;m:dl5000marker,1-5:特异引物验证,6-10:通用引物验证;
图5为甲醇诱导阳性转化子表达的表达产物sds-page电泳结果;1:蛋白marker,2-16:8-120h内每隔8h样品;
图6为甲醇诱导阳性转化子罐上表达的单宁酶tan1活力变化曲线;
图7为重组单宁酶tan1纯化结果sds-page电泳分析;m:蛋白marker1:未纯化蛋白,2:纯化蛋白;
图8为重组单宁酶的最适反应温度曲线;a:tan1,b:tan2;
图9为重组单宁酶的热稳定性曲线;a:tan1,b:tan2;
图10为重组单宁酶的最适反应ph曲线;a:tan1,b:tan2;
图11为重组单宁酶的ph稳定性曲线;a:tan1,b:tan2;
图12为重组单宁酶处理蓝莓果粉浸提液液相色谱图。1:ga,2:gc,3:egc,4:egcg,5:ec,6:gcg(1、2、3、5为非酯型儿茶素,4、6为酯型儿茶素);
图13为重组单宁酶酶法水解单宁酸产没食子酸曲线;
图14为重组单宁酶水解单宁酸产没食子酸反应薄层层析;a:tan1反应液薄层色谱,b:tan2反应液薄层色谱,1:单宁酸标品,2:没食子酸标品,3:单宁酸与没食子酸混标,4:反应0min样品,5:反应20min样品,6:反应40min样品,7:反应80min样品,8:反应120min样品。
具体实施方式
下面详细描述本发明的实施例,所述实施例的示例在附图中示出,其中自始至终相同或类似的标号表示相同或类似的元件或具有相同或类似功能的元件。下面通过参考附图描述的实施例是示例性的,旨在用于解释本发明,而不能理解为对本发明的限制。
下文的公开提供了许多不同的实施例或例子用来实现本发明的不同实施方式。为了简化本发明的公开,下文中对特定实施例或示例进行描述。当然,他们仅仅为示例,并且目的不在于限制本发明。此外,本发明提供的各种特定工艺和材料的例子,本领域普通技术人员可以意识到其他工艺的可应用性和/或其他材料的使用。除非另有说明,本发明的实施将采用本领域技术人员的能力范围之内的化学、分子生物学等领域的传统技术。另外,除非另有说明,在本文中,核酸以5’至3’的方向从左向右书写,氨基酸序列则以氨基端到羧基端的方向从左向右书写。
下面通过说明性的具体实施例对本发明进行描述,这些实施例并不以任何方式限制本发明的范围。特别说明的是:本发明所用到的试剂除特别说明外均有市售。
实验材料和试剂
1.菌株及载体:本发明所用黑曲霉(aspergillusnigersp.fj0118),于2019年05月16日保藏于中国典型培养物保藏中心,保藏编号为cctccno:m2019358,分类命名为aspergillusnigersp.fj0118黑曲霉fj0118,保藏地址为:中国.武汉.武汉大学。克隆载体pmd-19t购于takara公司,毕赤酵母表达载体ppic9k及菌株gs115购自于invitrogen公司。
2.酶类及其他生化试剂:内切酶及连接酶购自takara公司,罗丹宁购自aladddin公司,其它都为国产试剂(均可从普通生化试剂公司购买得到)。
3.溶液及培养基:
(1)pda培养基(用于保藏黑曲霉):去皮土豆200g,用蒸馏水煮至软而不烂后用八层纱布过滤取汁,加入20g葡萄糖及20g琼脂,定容至1l,分装至试管后121℃高压蒸汽灭菌20min,摆斜面冷却备用。
(2)lb培养基:蛋白胨10%,酵母粉5%,nacl10%,2%的琼脂,用于大肠杆菌的培养。
(3)ypd培养基:酵母粉10%,蛋白胨20%,葡萄糖20%,ph6.0,用于毕赤酵母的培养。
(4)md培养基;葡萄糖20g,琼脂粉20g,用0.9l蒸馏水溶解,121℃高压灭菌20min,使用时分别加入无菌的10×ynb100ml,以及0.02%生物素2ml,用于毕赤酵母的平板培养。
(5)bmgy培养基:酵母粉10g,蛋白胨20g,加0.7l蒸馏水溶解,121℃高压灭菌20min,冷却后在超净工作台内分别加入无菌的磷酸盐缓冲液100ml(1m,ph6.0),10×ynb100ml,10×甘油溶液100ml以及0.02%生物素溶液2ml,用于毕赤酵母的培养。
(6)bmmy培养基:将上述bmgy培养基中的碳源替换成为终浓度为0.5%的甲醇溶液100ml,用于毕赤酵母的诱导培养。
(7)罐上发酵培养基:85%磷酸80.1ml,0.093%caso4,1.82%k2so4,0.41%koh,1.5%mgso4·7h2o,4%甘油,0.5%酵母膏,0.5%蛋白胨,0.1%消泡油(v/v),溶解后定容至3l,注入发酵罐中,121℃高压灭菌20min,冷却后备用,用于毕赤酵母的培养。
需要说明的是:以下实施例中未作具体说明的分子生物学实验方法,均参照《分子克隆实验指南》(第三版)j.萨姆布鲁克一书中所列的具体方法进行,或者按照试剂盒和产品说明书进行。
实施例1黑曲霉基因组提取
接种黑曲霉(aspergillusnigersp.fj0118)菌株于pda斜面上,30℃,培养4d后,生理盐水制备孢子悬液,调至od600为2.0,孢子悬液接入50ml带玻璃珠的pda液体培养基中,于30℃、180rpm振荡培养24h,12000rpm离心收集菌体,菌体在液氮中快速研磨,直至研磨成细小白色粉末,后用plantgenomicdnaextractionkit按照其说明书操作,提取基因组dna。
实施例2单宁酶基因的pcr扩增
根据ncbi上公布的单宁酶基因(xm_001390374.1)序列信息,利用premier公司的primerpremier5设计引物,引物序列如下:
tan-f:5’-atgtccaagttcctcctctggacc-3’(seqidno:3);
tan-r:5’-ctagtagatcggaatgtcatacgcatcca-3’(seqidno:4);
以实施例1的黑曲霉基因组dna作为模板进行pcr扩增。反应参数为:95℃变性5min,94℃变性30sec,58~68℃梯度退火30sec,72℃延伸4min,28个循环后72℃保温10min,得到一段约2000bp片段,扩增后进行琼脂糖凝胶电泳验证,电泳结果如图1所示,将该片段回收后送伯瑞生物技术有限公司测序。
根据测序得到的核苷酸序列,设计上游和下游各两套tail-pcr特异性引物:设计方向为需要扩增的未知区域方向,qtan-f2/r2(seqidno:5和seqidno:7)的位置设计在qtan-f1/r1(seqidno:6和seqidno:8)的内侧。每两个引物之间的距离是60~100bp,引物长度一般22-30nt,退火温度在60-65℃。并将它们分别命名为qtan-f1,qtan-f2(上游特异性引物),qtan-r1,qtan-r2(下游特异性引物)见表1。
表1.单宁酶tail-pcr特异性引物
通过反向tail-pcr得到已知基因序列的侧翼序列,扩增得到的产物回收后送伯瑞生物技术有限公司测序。拼接后单宁酶tan1基因全长为1713bp,编码570个氨基酸和一个终止密码子。
使用signalp(http://www.cbs.dtu.dk/services/signalp)分析表明,拼接后单宁酶n端前17个氨基酸为信号肽。预测该基因所编码的成熟蛋白的理论分子量为61.14kda。
实施例3重组克隆质粒pmd19t-tan1的构建
1)单宁酶基因扩增产物与克隆载体连接
将实施例2得到的完整单宁酶基因重新设计引物进行扩增,得到的产物进行回收纯化与pmd19-tsimplevector(购自takara公司)连接构建重组克隆质粒。
拼接的单宁酶基因克隆所用引物如下:
wtan-f:5’-cctccttcagtctctcctttacctttg-3’(seqidno:9);
wtan-r:5’-gatgattccagttgtggctggtatta-3’(seqidno:10);
连接反应按照pmd19-tsimplevector连接试剂盒提供的说明书进行。在0.2mlpcr管中依次加入下列成分:
混匀后,瞬时离心,16℃下连接4h,获得连接产物。
2)大肠杆菌dh5α化学转化感受态细胞的制备
①使用lb平板培养基,用接种环挑取大肠杆菌(-20℃甘油保藏菌株),在平板上分级划线,于37℃倒置培养14-16h。
②从lb平板上挑取活化的e.colidh5α单菌落,接种于5mllb液体培养基中,37℃振荡培养12h。
③将上述培养物以1:100的比例接种于100ml的lb液体培养基中,37℃振荡培养至od600=0.5左右,放置冰上停止培养。
④取上述菌液1ml转入1.5ml离心管中,4000rpm,4℃离心10min,弃去上清。
⑤之后按competentcellpreparationkit(takara公司制备大肠感受态的试剂盒)说明书进行。
⑥冰上将感受态细胞分装成50μl/管,-80℃保存,获得感受态细胞dh5α。
3)克隆载体的转化及阳性转化子鉴定
从-80℃冰箱中取上述的感受态细胞dh5α,迅速冰浴解冻。取上述连接产物加入大肠杆菌感受态细胞dh5α中,轻轻混匀,冰浴30min,42℃水浴热击90s后,立即冰浴2min,加入1mllb液体培养基,37℃复苏2h。接着取200μl菌液涂布于含有amp抗性(终浓度为100μg/ml)的lb平板上,于37℃倒置培养12-16h。
挑取阳性单菌落接种到含有amp抗性(终浓度为100μg/ml)的5mllb液体培养基中,37℃、180rpm过夜培养。然后,通过菌液pcr验证阳性单菌落和测序分析表明克隆载体成功转化。
pcr验证的反应总体积为15μl,在0.2mlpcr管中依次加入下列成分:
混匀后瞬时离心,反应参数为:94℃变性2min;94℃变性30sec,60℃梯度退火30sec,72℃延伸1min30sec,28个循环后72℃保温2min。
菌液pcr结果如图2所示,综合测序结果显示黑曲霉单宁酶基因成功的连接到pmd19-t载体中。
实施例4重组表达载体ppic9k-tan1的构建
1)待插入的单宁酶基因片段的制备:
考虑到基因本身携带的信号肽序列会对毕赤酵母异源表达造成干扰,因此在设计引物时将上述预测的信号肽去除,并且加上酶切位点,引物序列如下:
mtan-f:5’-gagacctagggcgactctcgagcaggtctgta-3’(seqidno:11),5’分别引入保护碱基和avrⅱ酶切位点(下划线)
mtan-r:5’-attgcggccgcctagtagatcggaatgtcatacgcatc-3’(seqidno:12),5’分别引入保护碱基和notⅰ酶切位点(下划线)
以pmd19t-tan1重组质粒作为模板,反应总体积为50μl,在0.2mlpcr管中依次加入下列成分:
混匀后瞬时离心,反应参数为:95℃变性2min;95℃变性20sec,60℃梯度退火20sec,72℃延伸1min,28个循环后72℃保温5min。
用小量dna片段快速胶回收试剂盒(购自takara公司)回收上述去除信号肽的黑曲霉单宁酶基因,操作按产品说明书提供的步骤进行。
根据设计的上下游引物所带的酶切位点,选用avrⅱ酶和notⅰ酶对所回收纯化的单宁酶基因pcr产物进行双酶切,20μl酶切体系,如下:
混匀后瞬时离心,37℃酶切12h。酶切产物经1.0%琼脂糖凝胶电泳后,观察结果。将目的条带切下放入1.5ml离心管中。用小量dna片段快速胶回收试剂盒(购自takara公司),操作按产品说明书提供的步骤进行纯化回收。
2)毕赤酵母表达载体ppic9k(购自invitrogen公司)双酶切及其回收
双酶切采用20μl体系,如下:
混匀后瞬时离心,37℃酶切12h。酶切产物经1.0%琼脂糖凝胶电泳后,观察结果。将目的条带切下,放入1.5ml离心管中。使用小量dna片段快速胶回收试剂盒(购自takara公司),按产品说明书提供的步骤进行纯化回收。
3)酶切片段与表达载体的连接
将上述步骤1)获得的黑曲霉单宁酶基因,与上述步骤2)获得的表达载体ppic9k酶连,构建成表达载体ppic9k-tan1。连接体系如下:
混匀后瞬时离心,16℃下连接20min,获得连接产物。
4)连接产物转化大肠感受态细胞dh5α及阳性转化子验证
从-80℃冰箱中取上述实施例3的感受态细胞dh5α,迅速冰浴解冻。取上述步骤3)获得连接产物加入大肠杆菌感受态细胞dh5α中,轻轻混匀,冰浴30min,42℃水浴热击90s后,立即冰浴2min,加入1mllb液体培养基,37℃复苏2h。取200μl菌液涂布于含有amp抗性(终浓度为100μg/ml)的lb平板上,37℃倒置培养12~16h。
挑取阳性单菌落接种到含有amp抗性(终浓度为100μg/ml)的5mllb液体培养基中,37℃、180rpm过夜培养,通过菌液pcr验证阳性单菌落和测序分析表明表达载体成功转化。
反应总体积为15μl,在0.2mlpcr管中依次加入下列成分:
混匀后瞬时离心,反应参数为:94℃变性2min;94℃变性30sec,60℃梯度退火30sec,72℃延伸1min30sec,28个循环后72℃保温2min。
综合测序结果显示黑曲霉单宁酶基因成功的连接到ppic9k载体中。
实施例5表达载体ppic9k-tan1转化毕赤酵母gs115
1)将实施例4构建的表达载体ppic9k-tan1利用salⅰ单酶切线性化,线性化采用20μl体系,线性化加入下列成分:
混匀,瞬时离心,反应条件:37℃反应12h,反应完成后,用琼脂糖凝胶电泳检测线性化是否完全,结果如图3所示,线性化后为单一条带且电泳速度相比未线性化较慢,回收pcr产物,-20℃储存备用。
2)酵母菌株的转化及阳性克隆的筛选
将上述步骤1)线性化的表达载体ppic9k-tan1通过电击转化法转化至毕赤酵母gs115感受态细胞内,加入预冷的1mol/ld-山梨醇溶液,30℃复苏2h,之后取200μl菌液涂布于md固体培养基中初次筛选阳性克隆,30℃,倒置培养2~3d,观察单菌落生长情况。
之后从md平板上挑取单菌落接种于含有g148抗性(终浓度为2.5mg/ml)ypd平板内进行二次筛选多拷贝,30℃培养至长出菌落。挑取菌落接种于无抗液体ypd培养基中培养16~18h,取te/sds处理后菌液进行破壁pcr鉴定验证阳性转化子。
破壁菌液pcr体系如下:
混匀后瞬时离心,反应参数为:94℃变性2min;94℃变性30sec,60℃梯度退火30sec,72℃延伸1min30sec,28个循环后72℃保温2min。反应结束后,进行琼脂糖凝胶电泳进行分析鉴定,结果如图4所示,五个转化子都显示为阳性克隆,根据测序结果确定转化子模板。
实施例6诱导重组黑曲霉单宁酶tan1的高效表达
1)种子液制备
将实施例5筛选得到的重组菌株接种于ypd培养基中进行活化,30℃,250rpm培养16~18h。之后将活化的菌种按1:100的比例接种于ypd种子培养基内,30℃,250rpm培养16~18h,得到150ml的种子液。
2)罐上发酵
5l发酵罐,加入3l发酵培养基,121℃灭菌20min,冷却后加入微量元素ptm13.2ml,用氨水调整ph为5.2,按1:20比例接入种子液,发酵过程中温度控制在30℃,转速控制在600rpm左右。发酵具体分为以下三个阶段:
①甘油分批发酵阶段:发酵培养前16~24h(溶解氧(do)>40%;通气量:4l),直至培养基中甘油耗尽(表现为do迅速回升),补加甘油至菌体量达到180mg/ml。
②饥饿阶段:菌体达到所需密度后,停止补加甘油,溶氧(do)迅速回升,此时不添加碳源,保证菌体饥饿状态30min,以避免影响菌体对甲醇的利用。
③甲醇流加阶段:饥饿30min后,将发酵温度调整为至28℃,ph维持在5.0左右,开始甲醇诱导阶段。诱导前12h,补加甲醇(含1.2%ptm),补料速度为1.5ml/h添加;经12h诱导后,甲醇按3ml/h添加,持续96h。
发酵液上清用sds-page检测蛋白表达,结果如图5所示,随着发酵时间的延长,产酶量上升。
甘油补加阶段每隔8h取样测定生物量,甲醇诱导阶段每隔8h取样测定酶活力及生物量,结果如图6所示,生物量最高可达330mg/ml,罐上发酵诱导120h后,酶活力达到49.75u/ml,相比于摇瓶发酵,酶活力提高了53倍。zhong等利用毕赤酵母异源表达米曲霉的单宁酶基因,最大酶活为7000iu/l;selwal等利用青霉液态发酵产单宁酶,得到的酶活力为34.7u/ml。本申请获得的重组黑曲霉单宁酶酶活高于zhong和selwal等获得的单宁酶的酶活。
实施例7重组黑曲霉单宁酶的分离纯化
采用deaesepharosefastflow对上实施例6获得的重组黑曲霉单宁酶进行纯化,层析柱规格为1.6×20cm。分离纯化方法如下:
将上述实施例6获得的发酵上清液用0.22μm滤膜过滤,去除杂质,4℃冷藏备用。再经deaesepharosefastflow离子交换层析柱纯化,采用柠檬酸缓冲液(10mmol/l,ph5.0),nacl线性梯度(0.05-0.1mol/l),按1ml/min的流速进行洗脱,每管收集3ml,直至od280数值为零。
分别测定每管的蛋白含量及单宁酶活力,合并具有单宁酶酶活的洗脱液,用10kda的超滤膜超滤浓缩,进行sds-page验证分析纯化情况,结果见图7,纯化后为单一条带。
实施例8单宁酶的最适反应温度及热稳定性测定
在不同温度(30、40、50、60、70℃)条件下处理一段时间后,测定其酶活性(参考sharma(2000)的方法,将30℃条件下每分钟产生1μmol没食子酸所需要的酶量定义为一个酶活单位u)。测定热稳定性时,将酶液于不同温度下保温不同时间,测定其残余酶活性,以最高酶活力为100%,灭活10min的酶液为空白对照。结果如图8和图9所示。该重组黑曲霉单宁酶tan1的最适反应温度为50℃,在65℃可以保持70%以上的相对酶活。重组单宁酶tan1在低于50℃温度处理120min后,酶活力基本稳定不变,反而有上升的趋势。60℃保温120min依然保留50%以上的酶活力;tan2的最适反应温度为70℃,在70℃的水浴环境中处理10min后,单宁酶活力剩余10%左右,而在60℃和50℃的半衰期分别为8min和20min。以上结果表明,与文献中报道的丝状真菌来源的单宁酶相比,这两个单宁酶热稳定性良好,fuentes-garibay等报道来源于旱生黑曲霉(xerophilicaspergillusniger)菌株的单宁酶,在30℃保温120min后丧失50%的活力,表明不同来源的重组单宁酶温度稳定性差异显著。其中相比于tan2,tan1在高温下具有更好的耐热性。
实施例9单宁酶的最适反应ph及酸碱稳定性测定
将酶液用不同ph的缓冲液(1个ph单位为梯度)稀释到合适的倍数后,取0.25ml与等体积同ph的底物混匀,在30℃下测定单宁酶活力。以最高酶活力为100%,不同ph条件下灭活10min的酶液为空白对照,研究重组单宁酶的最适ph。测定酸碱稳定性时,将酶液用不同ph缓冲液稀释后,4℃放置24h,在标准情况下测定酶活力。测定结果如图10和图11所示。重组黑曲霉单宁酶tan1的最适反应ph为5.0。研究表明,大多数丝状真菌来源的单宁酶最适ph在酸性范围内(4.3~6.5),本研究结果与不同丝状真菌来源单宁酶的最适ph相似。特别注意的是,tan1在酸性至中性环境下较为稳定(ph2.0-7.0),在酸性条件下,放置24h,酶活基本没有丧失(残余相对酶活97.65%);tan2的最适反应ph为5.0,ph稳定性为3-7。相比于tan2,tan1在更低的ph环境中显示出更好的稳定性。
实施例10重组黑曲霉单宁酶tan1与其他来源单宁酶比较
据文献报道,不同来源的单宁酶的酶学性质存在差异。真菌和植物产单宁酶ph值稳定范围一般在4.0~6.0之间。细菌产单宁酶的最适宜ph值通常在中性范围,而真菌产单宁酶的稳定区间ph值为3.0~7.0。有关单宁酶酶学性质数据列于表1,对比表1中的大多数真菌来源的单宁酶,本发明的酶tan1在酸性条件下具有非常好的稳定性,可以在酸性食品的处理上有很大应用价值。
表1不同来源单宁酶酶学性质
*ng:文献中未给出数据
实施例11单宁酶在蓝莓果汁处理中的应用
鉴于本申请所得单宁酶在极酸性条件下的稳定性良好,所以研究在蓝莓果粉浸提液中添加该重组黑曲霉单宁酶tan1对其多酚含量的影响。
蓝莓多酚萃取:
取0.3g干燥蓝莓粉至具塞试管,按照体积比1:30加入萃取溶剂,用2mhcl将ph调为2.0,拧紧管塞,超声提取1h(频率400hz,温度45℃),后4000rpm离心5min分离上清与沉淀,上清即为萃取得到的蓝莓中的多酚物质。
重组黑曲霉单宁酶处理蓝莓果粉浸提液:
取以上4ml上清作为底物,加入1ml重组黑曲霉单宁酶tan1酶液,于50℃下处理2h,所得样品过膜后进行液相分析。结果如图12所示,经酶处理后,酯型儿茶素含量降低,非酯型儿茶素含量上升,表明本申请的单宁酶tan1可以应用于蓝莓果汁处理。因为,酯型儿茶素是果汁中苦涩味的来源,因此降低酯型儿茶素在蓝莓多酚中的比例,可以降低果汁中的涩味并提高果汁的储藏质量。
实施例12单宁酶在产没食子酸中的应用
用50ml蒸馏水在50℃的磁力搅拌器上溶解2g单宁酸10min,然后向溶解液中加入450u的单宁酶tan1或tan2,每隔20min取样检测没食子酸的生成,此过程中维持反应温度为50℃。并用薄层检测浸提液中单宁酸和没食子酸的变化,展开剂:正丁醇:乙酸:水(v/v/v)=2:2:1,显色剂:3%fecl3。
结果如图13所示,反应起始ph为5.0,随着反应进行,ph基本维持在3.0,在此条件下tan1的没食子酸转化率基本达到100%,但tan2的没食子酸转化率最高只达34.5%,图14所示与之对应,薄层分析结果显示tan1水解反应120min后基本没有单宁酸,其转换为没食子酸;但tan2水解反应120min后还是有大量单宁酸存在,所以相比于tan2,tan1在极酸性条件下更具有应用潜力。
在本说明书的描述中,参考术语“一个实施例”、“一些实施例”、“示例”、“具体示例”、或“一些示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不应理解为必须针对的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任何的一个或多个实施例或示例中以合适的方式结合。此外,本领域的技术人员可以将本说明书中描述的不同实施例或示例进行接合和组合。
尽管上面已经示出和描述了本发明的实施例,可以理解的是,上述实施例是示例性的,不能理解为对本发明的限制,本领域的普通技术人员在本发明的范围内可以对上述实施例进行变化、修改、替换和变型。
序列表
<110>集美大学
<120>一种耐酸型单宁酶及其基因和应用
<130>2019
<160>12
<170>siposequencelisting1.0
<210>1
<211>570
<212>prt
<213>人工序列(artificialsequence)
<400>1
metserlyspheleuleutrpthrvalseralaalaalaleualaarg
151015
alaalathrleugluglnvalcysthrvalalahisalalysalaala
202530
valproalaserglyleuvalglnglyvalvalthrasnproserser
354045
valthralavalprovaltyrasngluserthrserglyserasptyr
505560
tyrproalaglythrtyrasptyrcysasnilethrmetthrtyrser
65707580
hisalaglylysaspaspservalleuleuglnphetrpmetprothr
859095
proaspasppheglnasnargtrpvalserthrglyglypheglyphe
100105110
alaileasnvalalaserasnvalproalaglyleuprotyrglyala
115120125
valalaglyargthraspglyglypheglyserpheaspthrglnphe
130135140
serservaltyrproleuvalasnglythralaasntyraspalaleu
145150155160
tyrmetpheglytyrglnalahishisglumetserserileglylys
165170175
alaphethrlysasnphetyrsermetserasplysleutyralatyr
180185190
tyrglnglycyssergluglyglyarggluglytrpserglnvalgln
195200205
argpheprogluglutrpaspglyalavalileglyalaproalaile
210215220
argtyrglyglnglnglnvalasnhisleuvalproglnvalvalglu
225230235240
glnthrleuasptyrtyrprogluasncysglupheserglumetval
245250255
thrleuileileaspalacysaspgluleuaspglyarglysaspgly
260265270
valileglyargthraspleucysglnleuasnpheaspleulysser
275280285
thrileglylysprotyralacysasnalathrthrsermetglyval
290295300
thrthrproalaglnasnglythrilethralagluglyvalargval
305310315320
alaglnthrileleuaspglyleulysaspserglnglyargglnala
325330335
tyrphetrptyrargileglyserglupheseraspalagluthrthr
340345350
tyraspserthrthrglythrtyrglyileaspilesergluleugly
355360365
glyglutrpvalthrhisglyleuglnleuleuaspleuaspasnleu
370375380
serthrleuaspasnvalthrtyraspthrleuargasptrpmetleu
385390395400
gluglyleuglnargtyrgluaspvalleuglnthrthrtrpproasp
405410415
leuserargpheglnalaalaglyglylysvalilehistyrhisgly
420425430
gluseraspasnserileproproalaserservalargtyrpheglu
435440445
servalargaspthrmetpheproasnlysthrtyrasnalaserval
450455460
glualaleuasngluphetyrargleutyrleuvalproglyalaser
465470475480
hiscysasnthrasnserleuglnproasnglyprotyrproglnasn
485490495
serleuglyaspleumetasntrpvalglulysglyilegluproval
500505510
thrleuasnglythrvalleuserglyaspleugluglygluglugln
515520525
lysilecyssertrpproleuargproleutrplysasnasnglythr
530535540
glumetglucysvaltyraspglnlysserileaspthrtrpleutyr
545550555560
aspleuaspalatyraspileproiletyr
565570
<210>2
<211>1713
<212>dna
<213>人工序列(artificialsequence)
<400>2
atgtccaagttcctcctttggaccgtctccgcggcggccctcgcgcgcgccgcgactctc60
gagcaggtctgtaccgttgctcacgcaaaggccgccgtgcccgcatccggcctggtgcag120
ggtgtggtcaccaacccttccagtgtcactgcggttcccgtctacaatgaatccacctct180
ggatccgactactacccggccggaacctatgattactgcaatatcaccatgacttattct240
catgccggcaaggacgacagcgtcctgctgcaattctggatgcccactcccgatgacttc300
cagaaccgctgggtgtctaccggtggctttggctttgccatcaacgttgcctccaacgtg360
cctgctggtcttccctacggtgccgtcgccggccggaccgacggtggctttggcagcttc420
gacacccagttctcctctgtgtaccccttggtgaatggtaccgccaactacgatgccctg480
tacatgttcggctaccaggcccaccacgagatgtcctccatcggcaaggcgttcaccaag540
aacttctacagcatgtctgacaagctctatgcctactaccagggctgctccgagggtggt600
cgtgagggctggagtcaggtgcagcgcttccccgaggagtgggatggtgccgtcattggt660
gctcctgccatccgttatggccagcagcaggtgaaccacctggtgccccaggtcgtggag720
cagaccctcgactactaccctgagaactgcgagttctccgagatggtgactctgattatc780
gatgcctgtgatgaactcgatggccgtaaggatggcgtcatcggccgcaccgatctgtgc840
cagctcaacttcgacctcaagtccaccatcggtaagccttacgcctgcaacgccaccacc900
agcatgggtgtgaccaccccggcccagaacggcaccatcacggcggagggtgtgcgcgtc960
gctcagaccatcctagacggattgaaagactctcagggtcgccaggcctacttctggtac1020
cgcattggctcggagttcagcgatgcagagactacctacgactcgaccaccggcacctac1080
ggtatcgacatctccgagctgggtggtgagtgggtgactcacggactgcagctgctggac1140
ctcgacaacctctccactctggacaacgtcacctacgacaccctgcgtgactggatgctc1200
gagggtctccagcgctacgaggatgtcctgcagaccacctggcccgacctgtcccgcttc1260
caggctgcaggtggcaaggtcatccactaccacggtgaatcggacaacagcatccctccc1320
gcgtcttcggtgcgctacttcgagtccgtccgcgacactatgttccccaacaagacctac1380
aacgcctcggttgaagccttgaacgagttctaccgtctgtacctggttcccggtgcctcc1440
cactgcaacaccaactccctgcagcccaacggaccttacccccagaactccctcggcgac1500
ctgatgaactgggtcgagaagggcattgagcctgtgactctgaacggcaccgtgctgagc1560
ggcgatctcgagggagaggagcagaagatctgctcgtggcctctgcgtcctctgtggaag1620
aacaacggcacggagatggagtgcgtgtacgaccagaagtcgattgacacgtggttgtat1680
gatctggatgcgtatgacattccgatctactag1713
<210>3
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>3
atgtccaagttcctcctctggacc24
<210>4
<211>29
<212>dna
<213>人工序列(artificialsequence)
<400>4
ctagtagatcggaatgtcatacgcatcca29
<210>5
<211>27
<212>dna
<213>人工序列(artificialsequence)
<400>5
gtccttgccggcatgagaataagtcat27
<210>6
<211>27
<212>dna
<213>人工序列(artificialsequence)
<400>6
gtccttgccggcatgagaataagtcat27
<210>7
<211>26
<212>dna
<213>人工序列(artificialsequence)
<400>7
agttctaccgtctgtacctggttccc26
<210>8
<211>27
<212>dna
<213>人工序列(artificialsequence)
<400>8
atctcgagggagaggagcagaagatct27
<210>9
<211>27
<212>dna
<213>人工序列(artificialsequence)
<400>9
cctccttcagtctctcctttacctttg27
<210>10
<211>26
<212>dna
<213>人工序列(artificialsequence)
<400>10
gatgattccagttgtggctggtatta26
<210>11
<211>32
<212>dna
<213>人工序列(artificialsequence)
<400>11
gagacctagggcgactctcgagcaggtctgta32
<210>12
<211>38
<212>dna
<213>人工序列(artificialsequence)
<400>12
attgcggccgcctagtagatcggaatgtcatacgcatc38
- 还没有人留言评论。精彩留言会获得点赞!