一种超浸润表面及其制备方法和应用与流程
[0001]
本发明涉及高分子材料领域,具体涉及一种超浸润表面及其制备方法和应用。
背景技术:
[0002]
固体材料表面的润湿性通常是由固体表面的化学性质和微观结构决定的。具有超润湿性能的聚合物表面在液体分离,自清洁,防雾,液体输送,功能性聚合物薄膜,印刷和粘接等方面有重要用途。
[0003]
目前,已有众多学者对聚合物表面改性进行了深入研究,但超浸润材料方面的研究得到的大部分是超疏水材料,超亲水材料的报道十分少,显然超亲水聚合物较难制备。通过传统的接枝方法,例如atrp,电晕法,等离子体处理,紫外光等方法得到的聚合物表面只能是亲水表面却不是超亲水表面。
[0004]
超双亲表面比超亲水和超亲油表面更难制备,该表面要求水和油均能在很短时间内浸润材料表面。1997年,wang首次报道了双亲材料(wang,r.;hashimoto,k.;fujishima,a.;chikuni,m.;kojima,e.;kitamura,a.;shimohigoshi,m.;watanabe,t.,light-induced amphiphilic surfaces.nature 1997,388(6641),431-432.),它是通过将二氧化钛使用紫外光诱导在固体基材上制备出双亲表面,该表面具有防污能力。在此之后,人们利用逐层组装,静电纺丝,蚀刻,等离子体处理,浸涂,相分离和模板方法制备出多种超浸润聚合物表面。然而,通过上述方法制备的超浸润聚合物表面必须复合无机颗粒,在柔性制品中固体颗粒的脆性限制了材料的应用。因此,到目前为止,现有方法仍然不可能在不使用无机颗粒的情况下制备超双亲聚合物表面。
技术实现要素:
[0005]
本发明提供了一种亲水亲油性效果较好、持久稳定的超浸润聚丙烯表面,该产品的制备方法简便,易于工业化。其特点是将表面具有微纳结构的聚丙烯表面与有机酸及有机酸衍生物、乙烯基硅烷等亲水性单体在微波辐照下进行接枝反应,不添加引发剂以及辅助单体,得到表面富含有机酸及有机酸衍生物的亲水表面;还可以在此之后将其与碱反应,从而得到了表面富含有机酸盐的更亲水甚至超亲水的表面;还可以在表面富含有亲水性侧基的超浸润表面进一步接枝乙烯基硅油、苯乙烯等亲油性单体,可进一步提高亲水表面的亲油性。经本发明改性后的这种浸润表面,将具备超亲水、超亲油或者超双亲的超浸润性质。
[0006]
本发明的目的之一是提供一种超浸润表面。
[0007]
本发明所述的一种超浸润表面,为具有微纳结构的聚丙烯表面,所述聚丙烯表面接枝有亲水性侧基;或者同时接枝亲油性侧基和亲水性侧基;所述超浸润表面不含有引发剂残留物。
[0008]
以上所述具有微纳结构的聚丙烯表面,其所述微纳结构为具有微米或纳米尺度特征尺寸、按照特定方式排布的功能结构,所述的功能结构包括孔状结构或其他形状结构;通
常这个微纳结构的尺寸在1nm~100μm。
[0009]
所述的微纳结构的聚丙烯表面来自现有技术各种表面带有微纳结构的聚丙烯。
[0010]
以上所述的亲水性侧基优选为含有选自氧、硫、氮、硅和卤素及其组合的杂原子或其取代基并且含有碳碳双键的单体侧基;所述亲水性侧基的单体更优选为有机酸、有机酸的衍生物、乙烯基硅烷中的至少一种。
[0011]
以上所述有机酸的衍生物包括有机酸的酸酐、酯、盐中的至少一种。所述的有机酸包括但不限于羧酸、磺酸、亚磺酸、硫羧酸等。
[0012]
以上所述的亲水性侧基的单体进一步优选包括马来酸酐、马来酸酐衍生物、(甲基)丙烯酸、(甲基)丙烯酸衍生物(例如甲基丙烯酸缩水甘油酯)、乙酸乙烯酯、烯基磺酸及其衍生物(例如2-丙烯酰胺-2-甲基丙磺酸、丙烯磺酸、乙烯基苯磺酸、乙烯基磺酸等)、对苯乙烯甲酸、对苯乙烯乙酸、衣康酸、油酸、花生烯酸及其组合以及它们的成盐形式;和/或包括乙烯基硅烷;
[0013]
以上所述乙烯基硅烷为式(1)所示的化合物中的一种或多种:
[0014]
ch2=ch2(ch2)nsix3ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
式(1)
[0015]
其中n=0~3,x为氯基、甲氧基、乙氧基、甲氧基、乙氧基、乙酰氧基中的至少一种;
[0016]
所述乙烯基硅烷进一步优选为乙烯基三甲氧基硅烷、乙烯基三乙氧基硅烷中的至少一种。
[0017]
以上所述的亲油性侧基优选为乙烯基硅油侧基、苯乙烯侧基。
[0018]
以上所述的乙烯基硅油优选为端乙烯基硅油和高乙烯基硅油,进一步优选为甲基乙烯基硅油、乙烯基含氢硅油、二乙烯基硅油中的至少一种。
[0019]
本发明所述的超浸润表面水接触角比未改性处理前的聚丙烯表面水接触角小。所述的超浸润表面的水接触角优选小于90
°
,最优选为0
°
。也就是说本发明的超浸润表面在将聚丙烯表面进行接枝亲水基团后,其表面达到了亲水的效果。同时,聚丙烯表面接枝乙烯基硅油进行亲油改性后,其亲油性也进一步提高相比未亲油改性的聚丙烯表面以及仅亲水改性的聚丙烯表面都有所提高。
[0020]
本发明的目的之二是提供一种超浸润表面的制备方法。
[0021]
本发明所述的一种超浸润表面的制备方法,包括:
[0022]
将包括所述亲水性侧基的单体在内的组分、或者将包括所述亲水性侧基的单体和所述亲油性侧基的单体在内的组分,与聚丙烯表面在不加接枝引发剂的情况下使用微波辐照进行接枝反应而得所述超浸润表面;
[0023]
其中当组分不包括亲油性侧基的单体时,任选加入无机微波吸收介质;当组分包括亲油性侧基的单体时,加入无机微波吸收介质;
[0024]
或者当以上所述方法中所述亲水性性侧基的单体为有机酸或其酸酐或其酯中的至少一种时,包括将接枝反应后所得产物与碱反应的步骤(即所谓的盐化步骤)。
[0025]
本发明的制备方法中,微波辐照接枝反应可以只包括亲油侧基的微波辐照接枝反应;或者既包括亲油侧基的微波辐照接枝反应也包括亲水侧基的微波辐照接枝反应,两种侧基的微波辐照接枝反应可以同时发生,也可先后发生,顺序不限;所述的盐化步骤为任选的步骤,只要在聚丙烯表面接枝上有机酸或其酸酐或其酯中的至少一种侧基时即可进行,不限于其是否在亲油侧基的微波辐照接枝反应的前后,或其中(即在对亲水性性侧基的单
体为有机酸或其酸酐或其酯中的至少一种的接枝聚丙烯表面进行亲油性侧基微波辐照接枝的同时也可以加入碱对上述亲水侧基进行盐化)。
[0026]
本发明的制备方法,具体可包括以下方案中的任一种:
[0027]
方案一、包括将所述聚丙烯表面与所述亲水性侧基单体和/或其溶解在溶剂中的溶液接触混合,其中任选加入无机微波吸收介质;之后将所得的混合物在不加接枝引发剂的情况下微波辐照接枝;或者在混合物中还包含亲油性侧基的单体和/或其溶解在溶剂中的溶液,以及无机微波吸收介质;
[0028]
方案二、包括将所述聚丙烯表面与所述亲水性侧基单体和/或其溶解在溶剂中的溶液接触混合,其中任选加入无机微波吸收介质;之后将所得的混合物在不加接枝引发剂的情况下微波辐照接枝;然后将所得接枝产物与所述亲油性侧基单体和/或其溶解在溶剂中的溶液以及无机微波吸收介质混合,在不加接枝引发剂的情况下微波辐照接枝;
[0029]
方案三、包括将所述聚丙烯表面与所述亲油性侧基单体和/或其溶解在溶剂中的溶液以及无机微波吸收介质接触混合,之后将所得的混合物在不加接枝引发剂的情况下微波辐照接枝;然后将所得接枝产物与所述亲水性侧基单体和/或其溶解在溶剂中的溶液混合,在不加接枝引发剂的情况下微波辐照接枝;
[0030]
方案四、在以上所述三个方案的任一个基础上,当所述亲水性性侧基的单体为有机酸或其酸酐或其酯中的至少一种时,还包括将接枝了有机酸或其酸酐或其酯中的至少一种侧基的聚丙烯表面与碱和/或碱的水溶液接触混合的步骤(即所谓的盐化步骤)。
[0031]
根据以上制备方法,本发明所述超浸润表面中是不含有引发剂残留物。
[0032]
本发明的制备方法中所述的不添加接枝引发剂,其中所述接枝引发剂是指本领域现有技术已有常用于引发单体的聚合反应(包括接枝反应)的物质,例如自由基型引发剂,包括过氧化物引发剂和偶氮引发剂及氧化还原引发剂等。过氧化物引发剂又可分为有机过氧化物引发剂(例如过氧化二异丙苯)和无机过氧化物引发剂。尤其是指各种用于聚丙烯接枝功能单体的引发剂,例如过氧化二异丙苯等。现有技术的接枝方法中为了聚丙烯能与单体接枝,靠引发剂让聚丙烯的叔碳脱氢,但引发剂实际上不仅可以脱氢,还造成聚丙烯大量的β断链反应,也就是反应太剧烈,不可控。从而影响接枝聚丙烯的力学性能。本发明的制备方法无须添加引发剂即可在聚丙烯表面上接枝有机酸、有机酸衍生物、乙烯基硅烷、乙烯基硅油、苯乙烯等侧基。本发明所得到的超浸润表面,不含引发剂残留物,保证了聚丙烯表面的力学性能不受影响。
[0033]
更具体地,
[0034]
本发明的制备方法中,所述的聚丙烯表面可以为现有技术中已有的具有微纳结构的聚丙烯表面。所述聚丙烯表面的微纳结构的尺寸为1nm~100μm。
[0035]
本发明所述的制备方法中,所采用的聚丙烯表面可以为任意具有微纳结构的聚丙烯表面,其制备也采用现有技术已有的制备方法。具体的带有微纳结构的聚丙烯表面,例如可采用现有技术中已有的各种聚丙烯的微孔表面,优选采用热致相分离工艺制备的聚丙烯微孔平面。还可以采用现有技术的光刻技术、飞秒激光加工技术、等离子刻蚀技术、静电纺丝法、纳米压印、纳米铸造和超精密微铣技术等实现聚丙烯表面微纳结构的加工,具体比如使用表面具有微纳结构的金属模具,在聚丙烯表面压出微纳结构。或者使用电弧等方式在聚丙烯表面制备出微纳结构等。
[0036]
本发明的制备方法中,所述的亲水性侧基单体可采用现有技术中已有的各种亲水性单体,优选为含有选自氧、硫、氮、硅和卤素及其组合的杂原子或其取代基并且含有碳碳双键的单体,更优选包括有机酸、有机酸的衍生物、乙烯基硅烷中的至少一种。
[0037]
以上所述有机酸的衍生物包括有机酸的酸酐、酯、盐中的至少一种。所述的有机酸包括但不限于羧酸、磺酸、亚磺酸、硫羧酸等。
[0038]
以上所述有机酸和有机酸的衍生物优选选自马来酸酐、马来酸酐衍生物、(甲基)丙烯酸、(甲基)丙烯酸衍生物(例如甲基丙烯酸缩水甘油酯)、乙酸乙烯酯、烯基磺酸及其衍生物(例如2-丙烯酰胺-2-甲基丙磺酸、丙烯磺酸、乙烯基苯磺酸、乙烯基磺酸等)、对苯乙烯甲酸、对苯乙烯乙酸、衣康酸、油酸、花生烯酸及其组合以及它们的成盐形式;最优选马来酸酐、马来酸酐衍生物、(甲基)丙烯酸、(甲基)丙烯酸衍生物及其组合以及它们的成盐形式;最最优选马来酸酐及其成盐形式。
[0039]
以上所述的乙烯基硅烷优选为式(1)所示的化合物中的一种或多种:
[0040]
ch2=ch2(ch2)nsix3ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
式(1)
[0041]
其中n=0~3,x为氯基、甲氧基、乙氧基、甲氧基、乙氧基、乙酰氧基中的至少一种;所述乙烯基硅烷进一步优选为乙烯基三甲氧基硅烷、乙烯基三乙氧基硅烷中的至少一种。
[0042]
以上所述亲水性侧基的单体用量为所述聚丙烯表面用量的0.1~10%wt;优选为1~8%wt。
[0043]
以上所述亲水性侧基的单体既可以与聚丙烯表面或接枝亲油单体的聚丙烯表面直接接触混合,也可以为了混合效果更好采用亲水性侧基的单体的溶液进行混合。其溶剂的用量只要能够溶解所述单体形成溶液即可,优选亲水性侧基的单体溶液中所述单体与其溶剂的重量比为(0.1~100):100,优选(0.5~50):100,更优选(1~30):100。所述单体溶液的用量优选可以完全覆盖聚丙烯表面,更便于两者的充分接触混合。
[0044]
所述溶解亲水性侧基单体的溶剂选自水、有机溶剂中的至少一种;优选包括醇、酮、酯、水中至少一种,更优选丙酮或乙醇。
[0045]
本发明的制备方法中,所述亲油性侧基的单体可采用现有技术中已有的各种亲油性单体,优选包括乙烯基硅油、苯乙烯中的至少一种。其中所述乙烯基硅油优选为端乙烯基硅油和高乙烯基硅油,更优选甲基乙烯基硅油、乙烯基含氢硅油、二乙烯基硅油中的至少一种。
[0046]
所述亲油性侧基的单体用量为所述聚丙烯表面用量的0.1~30%wt;优选为1~20%wt。
[0047]
以上所述亲油性侧基的单体既可以与聚丙烯表面或接枝亲水单体的聚丙烯表面直接接触混合,也可以为了混合效果更好采用亲油性侧基的单体的溶液进行混合。其溶剂的用量只要能够溶解所述单体形成溶液即可,优选所述亲油性侧基单体与溶剂的重量比可以在(0.1~100):100,优选(0.5~50):100,更优选(1~30):100。所述单体溶液的用量优选可以完全覆盖所述聚丙烯表面,更便于两者的充分接触混合。
[0048]
所述溶解亲油性侧基单体的溶剂选自水、有机溶剂中的至少一种;优选包括醇、酮、酯、水中至少一种,更优选丙酮或乙醇。
[0049]
本发明的制备方法中,当聚丙烯表面只接枝亲水性侧基时,其单体与聚丙烯表面的混合物中可以不加入无机微波吸收介质,优选加入无机微波吸收介质以提高接枝效率。
当聚丙烯表面需要接枝亲油性侧基时,由于所述亲油性侧基的单体普遍在微波下升温不超过200℃,不能很好的进行接枝反应。因此必须加入无机微波吸收介质以促进微波下的接枝反应。
[0050]
以上所述无机微波吸收介质可采用现有技术中的各种可以吸收微波的无机物,优选包括金属氢氧化物、金属盐、金属氧化物、石墨类材料、铁电类材料、黄铜矿、电解石中的至少一种。
[0051]
所述金属氢氧化物为氢氧化钾、氢氧化钡、氢氧化钠、氢氧化锂、氢氧化锶、氢氧化钙、氢氧化铁、氢氧化亚铁、氢氧化锌、氢氧化镁、氢氧化钴、氢氧化金、氢氧化铝、氢氧化铜、氢氧化铍、稀土氢氧化物中的至少一种;所述金属盐选自硝酸铵、硝酸钾、硝酸钠、硝酸钡、硝酸钙、硝酸镁、硝酸铝、硝酸锰、硝酸锌、硝酸铁、硝酸亚铁、硝酸铜、硝酸银、氯化铵、氯化钾、氯化钠、氯化钡、氯化钙、氯化镁、氯化铝、氯化锰、氯化锌、氯化铁、氯化亚铁、氯化铜、硫酸铵、硫酸钾、硫酸钠、硫酸钙、硫酸镁、硫酸铝、硫酸锰、硫酸锌、硫酸铁、硫酸亚铁、硫酸铜、硫酸银、碳酸铵、碳酸钾、碳酸钠、碳酸镁、碳酸钙、碳酸钡、磷酸二氢钾、钛酸钡、钛酸锶、钛酸铜钙中的至少一种;所述金属氧化物选自三氧化二铁、四氧化三铁中的至少一种;所述石墨类材料选自炭黑、石墨粉、石墨烯、碳纳米管、活性炭中的至少一种。
[0052]
所述无机微波吸收介质的单次用量为聚丙烯表面用量的0.1~10%wt;优选为1~8%wt。所述无机微波吸收介质的单次用量,是指本发明的制备方法中包括的一次或若干次微波辐照,如果其中加入无机微波吸收介质,则其在单次微波辐照时的加入量。
[0053]
以上所述无机微波吸收介质既可以直接加入与聚丙烯表面或接枝聚丙烯表面接触混合,也可以为了混合效果更好,采用加入溶解或分散于溶剂得到无机微波吸收介质溶液或分散液进行接触混合。为了使得无机微波吸收介质更好的分散混合在(接枝)聚丙烯表面上,优选无机微波吸收介质与(接枝)聚丙烯表面的混合和与其他组分比如单体的混合分步进行,即(接枝)聚丙烯表面可以单独和单体组分混合烘干,然后烘干后的混合物再与无机微波吸收介质或其溶液或其分散液至少之一混合。
[0054]
所述溶解或分散微波吸收介质的溶剂用量只要能够溶解无机微波吸收介质形成无机微波吸收介质溶液、或是能够使得无机微波吸收介质充分均匀分散形成分散液即可。所述无机微波吸收介质溶液或分散液中溶剂与无机微波吸收介质的重量比优选可以在(0.1~100):100,更优选(0.5~50):100,最优选(1~30):100。
[0055]
所述无机微波吸收介质溶液或分散液的用量优选可以完全覆盖包括(接枝)聚丙烯表面在内的原料混合物,更便于原料的充分接触混合和反应。
[0056]
所述无机微波吸收介质溶液或分散液中溶剂选自水、有机溶剂中的至少一种;优选包括醇、酮、酯、水中至少一种,更优选醇、水。
[0057]
为了保障无机微波吸收介质能够与溶剂形成充分分散稳定的分散液,可以在所述无机微波吸收介质分散液中加入现有技术中通常的表面活性剂。一般可以选用聚氧乙烯型、多元醇型等表面活性剂,用量通常为无机微波吸收介质的0.1~100%wt。
[0058]
本发明的制备方法的所述盐化步骤中,碱选自现有技术中可以使得聚丙烯表面接枝的有机酸侧基、其酸酐侧基、其酯侧基的任一种盐化的碱即可;优选为氢氧化物。
[0059]
以上所述氢氧化物优选为金属氢氧化物和氨水中的至少一种;其中金属氢氧化物优选为氢氧化钠、氢氧化钾、氢氧化钡、氢氧化锂、氢氧化锶、氢氧化钙、氢氧化铁、氢氧化亚
铁、氢氧化锌、氢氧化镁、氢氧化钴、氢氧化金、氢氧化铝、氢氧化铜、氢氧化铍、氨水、稀土氢氧化物中的一种或几种,优选氢氧化钠、氢氧化钾、氢氧化钡、氢氧化锂、氢氧化锶、氢氧化钙中的一种或几种。
[0060]
以上所述氢氧化物用量为所述聚丙烯表面用量的0.1~10%wt;优选为1~8%wt。
[0061]
以上所述的碱与接枝聚丙烯表面的接触混合,可以选择直接加入碱进行接触混合,也可以为了便于混合充分,优选以碱的水溶液形式进行充分混合。其中溶解碱的水的用量也是只要能够溶解碱形成水溶液的即可。碱水溶液中水与碱的重量比可以优选在(0.1~100):100,更优选(0.5~50):100,最优选(1~30):100。所述碱的水溶液的量优选可以完全覆盖接枝聚丙烯表面,更便于两者的充分接触混合和反应。
[0062]
以上所述碱和/或碱水溶液与接枝聚丙烯表面充分混合并同时反应,即为通常的酸碱反应,其反应时间无特殊要求,至充分反应即可。一般是在碱和/或水溶液添加完毕后进一步接触混合同时反应一段时间即可,比如可以在30分钟以内,优选5-10分钟。其反应温度及压力都无限定,一般为常温常压。
[0063]
本发明的制备方法中,所述微波辐照的辐照功率为100w~2000w,优选为500~1000w,更优选600w~800w;辐射时间为1s~120min,优选1min~30min,再优选3min~10min。所述的微波辐照采用现有技术中已有的各种微波反应器中进行。
[0064]
所述的微波辐照可优选在惰性气氛下进行。所述惰性气氛可以采用现有技术中的惰性气体,优选包括氮气、氦气、氩气中的一种或几种,更优选氮气。
[0065]
本发明制备方法中,所述的混合优选在真空条件下进行。所述混合包括亲水性侧基的单体和/或其溶液与(接枝)聚丙烯表面的接触混合、亲油性侧基单体和/或其溶液与(接枝)聚丙烯表面的接触混合、接枝聚丙烯表面与碱和/或碱水溶液的接触混合等。
[0066]
对于本身带有微孔等微纳结构的聚丙烯表面来说,真空有利于接枝的单体和/或碱等其他组分与其接触混合的更充分,促进接枝单体和/或碱等其他组分进入到聚丙烯表面中,更有利于反应进行。
[0067]
本发明制备方法中所述的接触混合均可采用现有技术中已有的各种混合方法和设备,混合条件也为通常,只要能够实现各物料充分均匀混合即可;比如可以将包括所述单体等其他组分的原料或其溶液、分散液等涂覆、滴加、浸润、覆盖在所述聚丙烯表面实现接触混合。
[0068]
本发明制备方法中,所述单体与包括(接枝)聚丙烯表面在内的组分的混合物,优选在微波辐照前进行干燥处理。
[0069]
本发明制备方法中,所述微波辐照接枝后的产物优选使用溶剂清洗,以去除未反应的单体或者以及不参加反应的无机微波吸收介质,并优选在清洗后进一步干燥处理。
[0070]
以上所述的对微波辐照后的产物的清洗没有特殊限定,能够将残余的单体或者以及微波吸收介质去除即可,可采用通常的清洗方法。比如在微波后在高温的情况下立即使用体积超过聚丙烯表面的溶剂浸泡一定时间(比如5-15分钟),然后使用过滤装置去除多余水分;重复多次(比如2-6次)浸泡、过滤即得到清洗干净的超浸润表面。
[0071]
本发明制备方法中,所述盐化步骤的产物(即接枝反应产物与碱反应后的产物)优选使用溶剂清洗,去除未与接枝聚丙烯表面反应的碱,并优选在清洗后进一步干燥处理。
[0072]
以上所述对盐化后产物的清洗没有特殊限定,能够将残余的碱去除即可,可采用
通常的清洗方法。比如在盐化反应后立即使用体积超过接枝聚丙烯表面的溶剂浸泡一定时间(比如5-15分钟),然后使用过滤装置去除多余水分;重复多次(比如2-6次)浸泡、过滤即得到清洗干净的双亲性聚丙烯表面。
[0073]
本发明制备方法中,所述的清洗用溶剂选自水、有机溶剂中的至少一种;优选包括醇、酮、酯、水中至少一种,更优选醇、水。
[0074]
本发明制备方法中,所涉及的干燥处理可采用现有技术中各种常规干燥方法,包括但不限于如鼓风干燥、常温干燥等。优选的干燥温度不使聚丙烯产生熔融的温度为宜,例如不超过160℃。
[0075]
本发明的再一个目的是提供本发明所述的超浸润表面在粘接(例如塑料制品粘接等),喷涂(例如食品袋外包装的喷涂、汽车保险杠的喷涂等)等领域的应用。
[0076]
本发明的超浸润表面在能够同时亲水亲油,甚至可以达到超双亲。本发明采用将有机酸及有机酸衍生物等亲水性的单体与聚丙烯表面使用微波辐照在不添加引发剂的情况下进行接枝反应甚至包括进一步的盐化,因聚丙烯本身亲油,所以此时已形成了亲水亲油的表面;或者再进一步与乙烯基硅油等亲油性单体使用微波辐照在不添加引发剂的情况下进行接枝反应,来制备双亲的超浸润表面。不受任何理论约束,据信:聚丙烯在微波环境下是微波透明的(微波辐照下很少或者不吸收微波,因此在微波辐照下不发热)。所述作为接枝单体的有机酸及有机酸衍生物等单体在微波的条件下会吸收微波升温达200℃及以上,并产生自由基;同时高温也会引发附近聚丙烯分子链产生自由基,因此会与聚丙烯充分发生接枝反应,进而获得接枝聚丙烯表面。而同时不添加引发剂的微波接枝反应可以大幅避免添加引发剂接枝时的聚丙烯的β断链反应,不降低聚丙烯的分子量。进一步地,接枝有有机酸或其酸酐或其酯之一侧基的聚丙烯再与氢氧化物反应,可将接枝聚丙烯表面变成有机酸盐接枝聚丙烯表面,这将进一步提高聚丙烯的亲水性。为进一步提高聚丙烯表面的亲油性,需在聚丙烯上接枝乙烯基硅油等亲油性单体,乙烯基硅油等极性较低,其在微波辐照下吸收微波升温不能够达到很高温度(微波场中的温度升高至小于200℃),从而不能有效地引发附近聚丙烯分子链产生自由基,因此需要添加无机微波吸收介质来帮助聚丙烯产生自由基进而与乙烯基硅油单体发生接枝反应。无机微波吸收介质不与聚丙烯表面及单体反应,因此仅作为接枝反应热源,不影响聚丙烯表面性能。无机微波吸收介质的添加,对于不吸收微波的单体,可以帮助其接枝在聚丙烯上;对于本身吸收微波的单体,可以帮助提高其接枝效率。本发明利用微波的选择性加热,加热无机微波吸收介质,其微波下升温可达到的温度在200℃以上,可达到聚丙烯熔点附近,这个温度下聚丙烯不至于断链,但是聚丙烯叔碳可以脱氢,因此会发生接枝反应但不会造成断链反应。接枝乙烯基硅油等亲油性单体后,聚丙烯表面的亲油性进一步提高,所得聚丙烯表面具有双亲性。由于聚丙烯表面具有微纳结构,毛细作用会使亲水、亲油、双亲的表面变成超亲水、超亲油、超双亲的效果。
[0077]
本发明制备工艺简单,易操作。双亲改性方法适用于已经制备得到的所述聚丙烯表面,双亲性持久稳定,且无残留接枝单体、无残留碱、无残留引发剂等。设备简单,成本低,易于工业化。
[0078]
本发明的超浸润表面其接枝后聚丙烯分子量不下降、无残留单体、无引发剂残留、无色无味,亲水,亲油性均大幅提高且持久稳定。本发明制备工艺设备简单,易操作,成本低易于工业化,而且适合现有技术中已有的聚丙烯表面。
具体实施方式
[0079]
下面结合实施例,进一步说明本发明。本发明的范围不受这些实施例的限制,本发明的范围在附属的权利要求书中提出。
[0080]
接触角测试方法:
[0081]
采用德国kruss公司easy drop接触角测试仪,测量范围1~180
°
,分辨率
±
0.1
°
,采用动态接触角测量模式,每次固定体积为2μl的去离子水滴或者白油油滴,滴于聚丙烯表面上,取计算的初始接触角为聚丙烯表面的接触角测量值,平行测量6次,计算平均值。
[0082]
超浸润聚丙烯表面的接枝侧基测定方法:使用日本日立公司s4800扫描电镜的能谱配件对聚丙烯表面的接枝成分主要元素的含量进行测量;并通过接枝物分子式反推接枝物在聚丙烯表面的含量作为表面接枝率。(备注:由于是表面含量,故此比单体在原料中的含量偏高。)
[0083]
实施例及比较例所用原料:
[0084]
聚丙烯微纳表面制备方法:
[0085]
表面1:聚丙烯平板膜,天津膜天膜工程技术有限公司(平均孔径0.8μm,孔隙率80%),在膜的一面使用环氧树脂胶水封涂,另一面在液氮氛围中,使用leica cm3600冷冻切片机去除膜的皮层即得到聚丙烯微孔表面1,其表面的微孔平均尺寸为0.8μm。
[0086]
表面2:将聚丙烯树脂(t30s齐鲁石化,mi=3g/10min)注射出5cm
×
5cm厚度1mm的薄片,使用微小型超精密微细铣削机床,制备出具有微米结构的表面,具体为沿薄片表面横向纵向分别铣削,得到平均尺寸为0.5μm的表面微纳结构。
[0087]
表面3:将聚丙烯树脂(t30s齐鲁石化,mi=3g/10min)注射出5cm
×
5cm厚度1mm的的薄片,使用纳米压印机,制备出具有纳米结构的表面,具体为压印出平均尺寸为80nm小坑及凸起的表面微纳结构。
[0088]
马来酸酐(西陇科学股份有限公司),丙烯酸(国药集团化学试剂有限公司)、甲基丙烯酸(国药集团化学试剂有限公司)、2-丙烯酰胺-2-甲基丙磺酸(国药集团化学试剂有限公司)、氢氧化钠(西陇科学股份有限公司)、氢氧化钾(西陇科学股份有限公司)、氢氧化钙(西陇科学股份有限公司)、丙酮(西陇科学股份有限公司)、氯化钠(国药集团化学试剂有限公司)、乙烯基硅油(甲基乙烯基硅油,山东大易化工有限公司)、乙烯基含氢硅油(东京化成工业株式会社)、二乙烯基硅油(山东大易化工有限公司)、氯化钠(国药集团化学试剂有限公司)、氧化石墨烯(go)水溶液(南京吉仓纳米科技有限公司)、抗坏血酸(百灵威公司)、乙烯基三甲氧基硅烷(东京化成工业株式会社)、苯乙烯(国药集团化学试剂有限公司)。
[0089]
其他各种原料来自市售。
[0090]
实施例1:
[0091]
按聚丙烯表面(表面1)100质量份计,将马来酸酐(5质量份)溶解在丙酮(50质量份)中得到马来酸酐丙酮溶液;将氢氧化钠(5质量份)溶解在去离子水(50质量份)中得到氢氧化钠水溶液;将马来酸酐丙酮溶液在真空条件下加入到所述聚丙烯表面上充分接触混合,之后将混合物烘干(80℃鼓风干燥烘箱烘干)。将烘干后干燥的马来酸酐与聚丙烯表面的混合物在氮气的氛围下微波(功率700w)5min;微波完毕的产物在去离子水中浸泡10分钟并更换去离子水重复3遍以确保去除未参与接枝反应的马来酸酐单体,然后将其置于80℃鼓风干燥箱烘干得到干燥的马来酸酐接枝聚丙烯表面;将氢氧化钠水溶液在真空条件下与
烘干的马来酸酐接枝聚丙烯表面充分接触混合,氢氧化钠水溶液加入完后再混合并反应5分钟。反应完毕后使用去离子水按同上清洗步骤清洗反应产物然后将其置于80℃鼓风干燥箱烘干,得到接枝马来酸钠的聚丙烯表面。
[0092]
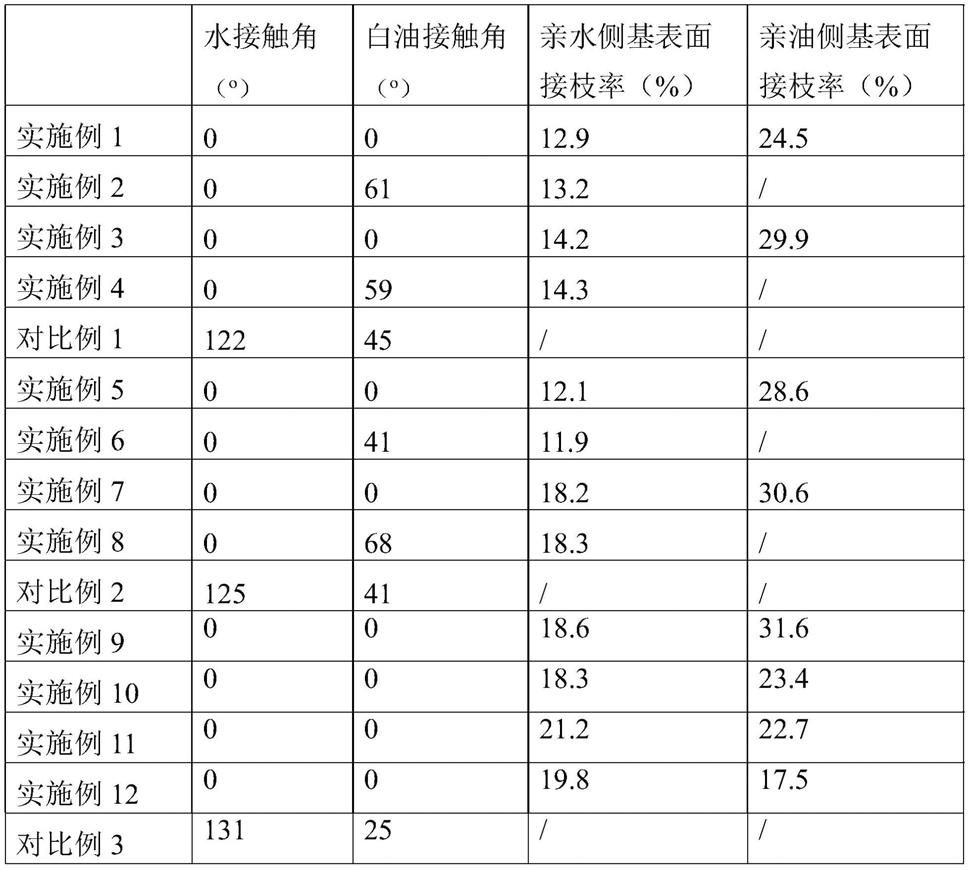
按所述聚丙烯表面100质量份计,将乙烯基硅油(5质量份)溶解在乙醇(50质量份)中得到乙烯基硅油乙醇溶液;将氯化钠(5质量份)溶解在去离子水(50质量份)中得到氯化钠水溶液;将乙烯基硅油乙醇溶液在真空条件下加入到上述所得的接枝马来酸钠聚丙烯表面上充分接触混合,之后将混合物烘干(80℃鼓风干燥烘箱烘干);将烘干后干燥的乙烯基硅油和接枝马来酸钠聚丙烯表面的混合物与氯化钠水溶液充分接触混合,之后将混合物烘干(80℃鼓风干燥烘箱烘干);将干燥的混合物在氮气的氛围下微波(功率700w)5min;微波完毕的物料在去离子水中浸泡10分钟并更换去离子水重复3遍以确保去除未参与接枝反应的乙烯基硅油单体以及氯化钠,然后将所得聚丙烯表面置于80℃鼓风干燥箱烘干,得到接枝马来酸钠和乙烯基硅油侧基的聚丙烯超浸润表面。所得超浸润表面的水、油接触角,表面接枝率数据见表1。
[0093]
实施例2:
[0094]
按聚丙烯表面(同实施例1)100质量份计,将马来酸酐(5质量份)溶解在丙酮(50质量份)中得到马来酸酐丙酮溶液;将氢氧化钠(5质量份)溶解在去离子水(50质量份)中得到氢氧化钠水溶液;将马来酸酐丙酮溶液在真空的条件下加入到所述聚丙烯表面上充分接触混合,之后将混合物烘干(80℃鼓风干燥烘箱烘干)。将烘干后干燥的马来酸酐与聚丙烯表面的混合物在氮气的氛围下微波(功率700w)5min;微波完毕的产物在去离子水中浸泡10分钟并更换去离子水重复3遍以确保去除未参与接枝反应的马来酸酐单体,然后其置于80℃鼓风干燥箱烘干得到干燥的马来酸酐接枝聚丙烯表面;将氢氧化钠水溶液在真空条件下加入至烘干的马来酸酐接枝聚丙烯表面充分接触混合,氢氧化钠水溶液加入完后再混合并反应5分钟。反应完毕后使用去离子水按同上清洗步骤清洗反应产物然后将其置于80℃鼓风干燥箱烘干,得到接枝马来酸钠侧基的聚丙烯超浸润表面。所得超浸润表面的水、油接触角、表面接枝率数据见表1。
[0095]
实施例3:
[0096]
按所述聚丙烯表面(同实施例1)100质量份计,将乙烯基硅油(9质量份)溶解在乙醇(50质量份)中得到乙烯基硅油乙醇溶液;将氯化钠(4质量份)溶解在去离子水(50质量份)中得到氯化钠水溶液;将乙烯基硅油乙醇溶液在真空条件下加入到所述聚丙烯表面上充分接触混合,之后将混合物烘干(80℃鼓风干燥烘箱烘干);将烘干后干燥的乙烯基硅油与聚丙烯表面的混合物与氯化钠水溶液充分接触混合,之后将混合物烘干(80℃鼓风干燥烘箱烘干);将干燥的混合物在氮气的氛围下微波(功率1000w)3min;微波完毕的物料在去离子水中浸泡10分钟并更换去离子水重复3遍以确保去除未参与接枝反应的乙烯基硅油单体以及氯化钠,然后将其置于80℃鼓风干燥箱烘干;得到接枝乙烯基硅油侧基的聚丙烯表面。
[0097]
按所述聚丙烯表面100质量份计,将丙烯酸(9质量份)溶解在丙酮(50质量份)中得到丙烯酸丙酮溶液;将氢氧化钾(6质量份)溶解在去离子水(50质量份)中得到氢氧化钾水溶液;将丙烯酸丙酮溶液在真空条件下加入到接枝聚丙烯表面上充分接触混合,之后将混合物烘干(80℃鼓风干燥烘箱烘干)。将烘干后干燥的丙烯酸与接枝聚丙烯表面的混合物在
氮气的氛围下微波(功率1000w)3min;微波完毕的产物在去离子水中浸泡10分钟并更换去离子水重复3遍以确保去除未参与接枝反应的丙烯酸单体,然后将其置于80℃鼓风干燥箱烘干得到干燥的接枝有丙烯酸和乙烯基硅油接枝聚丙烯表面;将氢氧化钾水溶液在真空条件下加入至烘干的接枝有丙烯酸和乙烯基硅油的聚丙烯表面充分接触混合,氢氧化钾水溶液加入完后再混合并反应5分钟。反应完毕后使用去离子水按同上清洗步骤清洗反应产物然后将其置于80℃鼓风干燥箱烘干,得到接枝有丙烯酸钾和乙烯基硅油侧基的聚丙烯超浸润表面。所得超浸润表面的水、油接触角、表面接枝率数据见表1。
[0098]
实施例4:
[0099]
按聚丙烯表面(同实施例1)100质量份计,将丙烯酸(9质量份)溶解在丙酮(50质量份)中得到丙烯酸丙酮溶液;将氢氧化钾(6质量份)溶解在去离子水(50质量份)中得到氢氧化钾水溶液;将丙烯酸丙酮溶液在真空条件下加入到所述聚丙烯表面上充分接触混合,之后将混合物烘干(80℃鼓风干燥烘箱烘干)。将烘干后干燥的丙烯酸与聚丙烯表面的混合物在氮气的氛围下微波(功率1000w)3min;微波完毕的产物在去离子水中浸泡10分钟并更换去离子水重复3遍以确保去除未参与接枝反应的丙烯酸单体,然后将其置于80℃鼓风干燥箱烘干得到干燥的丙烯酸接枝聚丙烯表面;将氢氧化钾水溶液在真空条件下加入至烘干的丙烯酸接枝的聚丙烯表面充分接触混合,氢氧化钾水溶液加入完后再混合并反应5分钟。反应完毕后使用去离子水按同上清洗步骤清洗反应产物然后将其置于80℃鼓风干燥箱烘干,得到接枝丙烯酸钾侧基的聚丙烯超浸润表面。所得超浸润表面的水、油接触角、表面接枝率数据见表1。
[0100]
对比例1:
[0101]
将聚丙烯表面(同实施例1)直接进行测试,聚丙烯表面的水、油接触角数据见表1。
[0102]
实施例5:
[0103]
按聚丙烯表面(表面2)100质量份计,将2-丙烯酰胺-2-甲基丙磺酸(10质量份)溶解在丙酮(50质量份)中得到2-丙烯酰胺-2-甲基丙磺酸丙酮溶液;将氢氧化钾(6质量份)溶解在去离子水(50质量份)中得到氢氧化钾水溶液;将2-丙烯酰胺-2-甲基丙磺酸丙酮溶液在真空条件下加入到所述聚丙烯表面上充分接触混合,之后将混合物烘干(80℃鼓风干燥烘箱烘干)。将烘干后干燥的2-丙烯酰胺-2-甲基丙磺酸与聚丙烯表面的混合物在氮气的氛围下微波(功率1000w)3min;微波完毕的产物在去离子水中浸泡10分钟并更换去离子水重复3遍以确保去除未参与接枝反应的2-丙烯酰胺-2-甲基丙磺酸单体,然后将其置于80℃鼓风干燥箱烘干得到干燥的2-丙烯酰胺-2-甲基丙磺酸接枝聚丙烯表面;将氢氧化钾水溶液在真空条件下与烘干的2-丙烯酰胺-2-甲基丙磺酸接枝的聚丙烯表面充分接触混合,氢氧化钾水溶液加入完后再混合并反应5分钟。反应完毕后使用去离子水按同上清洗步骤清洗反应产物然后将其置于80℃鼓风干燥箱烘干,得到接枝2-丙烯酰胺-2-甲基丙磺酸钠的聚丙烯表面。
[0104]
按所述聚丙烯表面100质量份计,将乙烯基含氢硅油(9质量份)溶解在乙醇(50质量份)中得到乙烯基含氢硅油乙醇溶液;将氯化钠(4质量份)溶解在去离子水(50质量份)中得到氯化钠水溶液;将乙烯基含氢硅油乙醇溶液在真空条件下加入到上述接枝2-丙烯酰胺-2-甲基丙磺酸钠的聚丙烯表面上充分接触混合,之后将混合物烘干(80℃鼓风干燥烘箱烘干);将烘干后干燥的乙烯基含氢硅油与接枝聚丙烯表面的混合物与氯化钠水溶液充分
接触混合,之后将混合物烘干(80℃鼓风干燥烘箱烘干);将干燥的混合物在氮气的氛围下微波(功率1000w)3min;微波完毕的物料在去离子水中浸泡10分钟并更换去离子水重复3遍以确保去除未参与接枝反应的乙烯基含氢硅油单体以及氯化钠,然后将其置于80℃鼓风干燥箱烘干;得到接枝2-丙烯酰胺-2-甲基丙磺酸钠和乙烯基含氢硅油侧基的超浸润表面。所得超浸润表面的水、油接触角、表面接枝率数据见表1。
[0105]
实施例6:
[0106]
按聚丙烯表面(同实施例5)100质量份计,将2-丙烯酰胺-2-甲基丙磺酸(10质量份)溶解在丙酮(50质量份)中得到2-丙烯酰胺-2-甲基丙磺酸丙酮溶液;将氢氧化钾(6质量份)溶解在去离子水(50质量份)中得到氢氧化钾水溶液;将2-丙烯酰胺-2-甲基丙磺酸丙酮溶液在真空条件下加入到聚丙烯表面上充分接触混合,之后将混合物烘干(80℃鼓风干燥烘箱烘干)。将烘干后干燥的2-丙烯酰胺-2-甲基丙磺酸与聚丙烯表面的混合物在氮气的氛围下微波(功率1000w)3min;微波完毕的产物在去离子水中浸泡10分钟并更换去离子水重复3遍以确保去除未参与接枝反应的2-丙烯酰胺-2-甲基丙磺酸单体,然后其置于80℃鼓风干燥箱烘干得到干燥的2-丙烯酰胺-2-甲基丙磺酸接枝聚丙烯表面;将氢氧化钾水溶液在真空条件下加入至烘干的2-丙烯酰胺-2-甲基丙磺酸接枝的聚丙烯表面充分接触混合,氢氧化钾水溶液加入完后再混合并反应5分钟。反应完毕后使用去离子水按同上清洗步骤清洗反应产物然后将其置于80℃鼓风干燥箱烘干,得到接枝2-丙烯酰胺-2-甲基丙磺酸钾侧基的超浸润表面。所得超浸润表面的水、油接触角、表面接枝率数据见表1。
[0107]
实施例7:
[0108]
按聚丙烯表面(同实施例5)100质量份计,将甲基丙烯酸(10质量份)溶解在丙酮(50质量份)中得到甲基丙烯酸丙酮溶液;将氢氧化钙(8质量份)溶解在去离子水(50质量份)中得到氢氧化钙水溶液;将甲基丙烯酸丙酮溶液在真空条件下加入到所述聚丙烯表面上充分接触混合,之后将混合物烘干(80℃鼓风干燥烘箱烘干)。将烘干后干燥的甲基丙烯酸与聚丙烯表面的混合物在氮气的氛围下微波(功率2000w)1min;微波完毕的产物在去离子水中浸泡10分钟并更换去离子水重复3遍以确保去除未参与接枝反应的甲基丙烯酸单体,然后将其置于80℃鼓风干燥箱烘干得到干燥的甲基丙烯酸接枝聚丙烯表面;将氢氧化钙水溶液在真空条件下加入至烘干的甲基丙烯酸接枝的聚丙烯表面充分接触混合,氢氧化钙水溶液加入完后再混合并反应5分钟。反应完毕后使用去离子水按同上清洗步骤清洗反应产物然后将其置于80℃鼓风干燥箱烘干,得到接枝甲基丙烯酸钙的聚丙烯表面。
[0109]
按所述聚丙烯表面100质量份计,将二乙烯基硅油(8质量份)溶解在乙醇(50质量份)中得到二乙烯基硅油乙醇溶液;将氯化钠(6质量份)溶解在去离子水(50质量份)中得到氯化钠水溶液;将二乙烯基硅油乙醇溶液在真空条件下加入到上述接枝甲基丙烯酸钾的聚丙烯表面上充分接触混合,之后将混合物烘干(80℃鼓风干燥烘箱烘干);将烘干后干燥的二乙烯基硅油与接枝聚丙烯表面的混合物与氯化钠水溶液充分接触混合,之后将混合物烘干(80℃鼓风干燥烘箱烘干);将干燥的混合物在氮气的氛围下微波(功率2000w)1min;微波完毕的物料在去离子水中浸泡10分钟并更换去离子水重复3遍以确保去除未参与接枝反应的二乙烯基硅油单体以及氯化钠,然后将其置于80℃鼓风干燥箱烘干;得到接枝甲基丙烯酸钙和二乙烯基硅油侧基的聚丙烯超浸润表面。所得超浸润表面的水、油接触角、表面接枝率数据见表1。
[0110]
实施例8:
[0111]
按聚丙烯表面(同实施例5)100质量份计,将甲基丙烯酸(10质量份)溶解在丙酮(50质量份)中得到甲基丙烯酸丙酮溶液;将氢氧化钙(8质量份)溶解在去离子水(50质量份)中得到氢氧化钙水溶液;将甲基丙烯酸丙酮溶液在真空条件下加入到所述聚丙烯表面上充分接触混合,之后将混合物烘干(80℃鼓风干燥烘箱烘干)。将烘干后干燥的甲基丙烯酸与聚丙烯表面的混合物在氮气的氛围下微波(功率2000w)1min;微波完毕的产物在去离子水中浸泡10分钟并更换去离子水重复3遍以确保去除未参与接枝反应的甲基丙烯酸单体,然后将其置于80℃鼓风干燥箱烘干得到干燥的甲基丙烯酸接枝聚丙烯表面;将氢氧化钙水溶液在真空条件下加入至烘干的甲基丙烯酸接枝的聚丙烯表面充分接触混合,氢氧化钙水溶液加入后再混合并反应5分钟。反应完毕后使用去离子水按同上清洗步骤清洗反应产物然后将其置于80℃鼓风干燥箱烘干,得到接枝甲基丙烯酸钙侧基的聚丙烯超浸润表面。所得超浸润表面的水、油接触角、表面接枝率数据见表1。
[0112]
对比例2:
[0113]
将聚丙烯表面(同实施例5)直接进行测试,聚丙烯表面的水、油接触角,数据见表1。
[0114]
实施例9:
[0115]
按聚丙烯表面(表面3)100质量份计,将甲基丙烯酸(6质量份)溶解在丙酮(50质量份)中得到甲基丙烯酸丙酮溶液;将甲基丙烯酸丙酮溶液在真空条件下加入到所述聚丙烯表面上充分接触混合,之后将混合物烘干(80℃鼓风干燥烘箱烘干)。将烘干后干燥的甲基丙烯酸与聚丙烯表面的混合物在氮气的氛围下微波(功率500w)30min;微波完毕的产物在去离子水中浸泡10分钟并更换去离子水重复3遍以确保去除未参与接枝反应的甲基丙烯酸单体,然后将其置于80℃鼓风干燥箱烘干得到干燥的甲基丙烯酸接枝聚丙烯表面。
[0116]
按所述聚丙烯表面100质量份计,将二乙烯基硅油(10质量份)溶解在乙醇(50质量份)中得到二乙烯基硅油乙醇溶液;将氯化钠(6质量份)溶解在去离子水(50质量份)中得到氯化钠水溶液;将二乙烯基硅油乙醇溶液在真空条件下加入到上述甲基丙烯酸接枝的聚丙烯表面上充分接触混合,之后将混合物烘干(80℃鼓风干燥烘箱烘干);将烘干后干燥的二乙烯基硅油和接枝聚丙烯表面的混合物与氯化钠水溶液充分接触混合,之后将混合物烘干(80℃鼓风干燥烘箱烘干);将干燥的混合物在氮气的氛围下微波(功率500w)30min;微波完毕的物料在去离子水中浸泡10分钟并更换去离子水重复3遍以确保去除未参与接枝反应的二乙烯基硅油单体以及氯化钠,然后将其置于80℃鼓风干燥箱烘干;得到接枝甲基丙烯酸和二乙烯基硅油的聚丙烯超浸润表面。所得超浸润表面的水、油接触角、表面接枝率数据见表1。
[0117]
实施例10:
[0118]
按聚丙烯表面(同实施例9)100质量份计,将甲基丙烯酸(1质量份)溶解在丙酮(50质量份)中得到甲基丙烯酸丙酮溶液;将甲基丙烯酸丙酮溶液在真空条件下加入到所述聚丙烯表面上充分接触混合,之后将混合物烘干(80℃鼓风干燥烘箱烘干)。将烘干后干燥的甲基丙烯酸与聚丙烯表面的混合物在氮气的氛围下微波(功率500w)30min;微波完毕的产物在去离子水中浸泡10分钟并更换去离子水重复3遍以确保去除未参与接枝反应的甲基丙烯酸单体,然后将其置于80℃鼓风干燥箱烘干得到干燥的甲基丙烯酸接枝聚丙烯表面。
[0119]
按所述聚丙烯表面100质量份计,将甲基乙烯基硅油(2质量份)溶解在乙醇(50质量份)中得到甲基乙烯基硅油乙醇溶液;将氧化石墨烯(go)水溶液(10质量份)、抗坏血酸(1质量份)溶解在去离子水(50质量份)中得到氧化石墨烯(go)分散液;将甲基乙烯基硅油乙醇溶液在真空条件下加入到上述甲基丙烯酸接枝聚丙烯表面上充分接触混合,之后将混合物烘干(80℃鼓风干燥烘箱烘干);将烘干后干燥的甲基乙烯基硅油和接枝聚丙烯表面混合物的粉料与氧化石墨烯(go)分散液充分接触混合,之后将混合物烘干(80℃鼓风干燥烘箱烘干),其中氧化石墨烯、抗坏血酸与去离子水混合形成氧化石墨烯分散液,氧化石墨烯分散液在与甲基乙烯基硅油和接枝聚丙烯表面的混合物混合后,在80℃烘干干燥时,抗坏血酸作为氧化石墨烯的还原剂将氧化石墨烯还原成石墨烯,石墨烯是后续微波辐照接枝的微波吸收介质;将干燥的混合物在氮气的氛围下微波(功率500w)30min;微波完毕的物料在去离子水中浸泡10分钟并更换去离子水重复3遍以确保去除未参与接枝反应的甲基乙烯基硅油单体以及氧化石墨烯,然后将其置于80℃鼓风干燥箱烘干;得到接枝甲基丙烯酸和甲基乙烯基硅油侧基的聚丙烯超浸润表面。所得超浸润表面的水、油接触角、表面接枝率数据见表1。
[0120]
实施例11:
[0121]
按聚丙烯表面(同实施例9)100质量份计,将甲基丙烯酸(7质量份)溶解在丙酮(50质量份)中得到甲基丙烯酸丙酮溶液;将甲基丙烯酸丙酮溶液在真空条件下加入到所述聚丙烯表面上充分接触混合,之后将混合物烘干(80℃鼓风干燥烘箱烘干)。将烘干后干燥的甲基丙烯酸与聚丙烯表面的混合物在氮气的氛围下微波(功率500w)30min;微波完毕的产物在去离子水中浸泡10分钟并更换去离子水重复3遍以确保去除未参与接枝反应的甲基丙烯酸单体,然后将其置于80℃鼓风干燥箱烘干得到干燥的甲基丙烯酸接枝聚丙烯表面。
[0122]
按所述聚丙烯表面100质量份计,将甲基乙烯基硅油(8质量份)溶解在乙醇(50质量份)中得到甲基乙烯基硅油乙醇溶液;将氧化石墨烯(go)水溶液(3质量份)、抗坏血酸(0.3质量份)溶解在去离子水(50质量份)中得到氧化石墨烯(go)分散液;将甲基乙烯基硅油乙醇溶液在真空条件下加入到上述甲基丙烯酸接枝聚丙烯表面上充分接触混合,之后将混合物烘干(80℃鼓风干燥烘箱烘干);将烘干后干燥的甲基乙烯基硅油和接枝聚丙烯表面的混合物与氧化石墨烯(go)分散液充分接触混合,之后将混合物烘干(80℃鼓风干燥烘箱烘干),其中氧化石墨烯、抗坏血酸与去离子水混合形成氧化石墨烯分散液,氧化石墨烯分散液在与甲基乙烯基硅油和接枝聚丙烯表面的混合物混合后,在80℃烘干干燥时,抗坏血酸作为氧化石墨烯的还原剂将氧化石墨烯还原成石墨烯,石墨烯是后续微波辐照接枝的微波吸收介质;将干燥的混合物在氮气的氛围下微波(功率500w)30min;微波完毕的物料在去离子水中浸泡10分钟并更换去离子水重复3遍以确保去除未参与接枝反应的甲基乙烯基硅油单体以及氧化石墨烯,然后将其置于80℃鼓风干燥箱烘干;得到接枝甲基丙烯酸和甲基乙烯基硅油的聚丙烯超浸润表面。所得超浸润表面的水、油接触角数据见表1。
[0123]
实施例12:
[0124]
按聚丙烯表面(同实施例9)100质量份计,将乙烯基三甲氧基硅烷(9质量份)溶解在乙醇(50质量份)中得到乙烯基三甲氧基硅烷乙醇溶液;将氯化钠(3质量份)溶解在去离子水(50质量份)中得到氯化钠水溶液;将乙烯基三甲氧基硅烷乙醇溶液在真空条件下加入到所述聚丙烯表面上充分接触混合,之后将混合物烘干(80℃鼓风干燥烘箱烘干)。将烘干
后干燥的乙烯基三甲氧基硅烷和聚丙烯表面混合物与氯化钠水溶液充分接触混合,之后将混合物烘干(80℃鼓风干燥烘箱烘干);将烘干后干燥的乙烯基三甲氧基硅烷与聚丙烯表面的混合物在氮气的氛围下微波(功率500w)30min;微波完毕的产物在去离子水中浸泡10分钟并更换去离子水重复3遍以确保去除未参与接枝反应的乙烯基三甲氧基硅烷单体和氯化钠,然后将其置于80℃鼓风干燥箱烘干得到干燥的乙烯基三甲氧基硅烷接枝聚丙烯表面。
[0125]
按所述聚丙烯表面100质量份计,将苯乙烯(8质量份)溶解在乙醇(50质量份)中得到苯乙烯乙醇溶液;将氧化石墨烯(go)水溶液(4质量份)、抗坏血酸(0.4质量份)溶解在去离子水(50质量份)中得到氧化石墨烯(go)分散液;将苯乙烯乙醇溶液在真空条件下加入到上述苯乙烯接枝聚丙烯表面上充分接触混合,之后将混合物烘干(80℃鼓风干燥烘箱烘干);将烘干后干燥的苯乙烯和接枝聚丙烯表面混合物的粉料与氧化石墨烯(go)分散液充分接触混合,之后将混合物烘干(80℃鼓风干燥烘箱烘干),其中氧化石墨烯、抗坏血酸与去离子水混合形成氧化石墨烯分散液,氧化石墨烯分散液在与苯乙烯和接枝聚丙烯表面的混合物混合后,在80℃烘干干燥时,抗坏血酸作为氧化石墨烯的还原剂将氧化石墨烯还原成石墨烯,石墨烯是后续微波辐照接枝的微波吸收介质;将干燥的混合物在氮气的氛围下微波(功率500w)30min;微波完毕的物料在去离子水中浸泡10分钟并更换去离子水重复3遍以确保去除未参与接枝反应的苯乙烯单体以及氧化石墨烯,然后将其置于80℃鼓风干燥箱烘干;得到接枝乙烯基三甲氧基硅烷和苯乙烯的双亲聚丙烯表面。所得双亲聚丙烯表面的水、油接触角、表面接枝率数据见表1。
[0126]
对比例3:
[0127]
将聚丙烯表面(同实施例9)直接进行测试,聚丙烯表面的水、油接触角,数据见表1。
[0128]
表1
[0129][0130]
从表1实施例中可以看出,经过本发明对聚丙烯表面进行亲水、亲油接枝改性后,相比纯聚丙烯表面,其水、油通量均大幅度提升,改性后聚丙烯表面均为超亲水、亲油,甚至有的改性后的聚丙烯表面达到超亲水和超亲油,说明聚丙烯表面双亲改性非常有效。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1