EMC3基因的应用及其定点敲除方法与流程
本发明属于生物技术领域,具体涉及emc3基因的应用及其定点敲除方法。
背景技术:
由致病微生物引起的各类传染病一直是影响全球养猪业高效发展的一大障碍。中国每年由于传染病造成的养猪经济损失预估高达400亿元,特别是自2018年以来,非洲猪瘟传入中国,重创了国内养猪业。中国是世界上最大的生猪养殖和猪肉消费国,疫情不仅影响生猪产业,也影响着相关国际贸易。如何有效预防和控制疾病发生一直是畜牧业关注的重点。尽管预防接种发挥了重要的防治作用,但始终未能完全控制和消灭传染病的流行。从长远来看,采用遗传学策略能够从遗传本质上提高猪对病原的抗性,所以开展抗病育种研究具有治本的功效。随着分子生物学技术的日趋成熟,目前抗病育种研究取得了一系列进展,如陆续发现干扰素基因、mx1、防御素、nramp1和mhc等候选基因可用于抗病育种。特别是crispr/cas9技术的兴起,结合体细胞核移植技术或受精卵显微注射技术,分别成功改变了猪cd163和anpep基因,制备了能有效抵抗猪蓝耳病毒(prrsv)或猪传染性胃肠炎病毒(tgev)侵染和复制的抗病猪,为开展基因组编辑抗病育种提供了重要参考。尽管有些致病微生物的抗病基因已被鉴定,但能感染猪的细菌或病毒的种类非常多,因而依然需要加强研究力度,开展持续性的抗病育种研究。
流行性乙型脑炎(je)是由乙型脑炎病毒(japaneseencephalitisvirus,jev)引起的一种以虫蚊为传播媒介的人畜共患传染病。jev主要在猪—蚊—人这一循环中传播,其中猪是jev重要的扩增贮存宿主,带毒蚊子通过叮咬将jev传播给人,人是终末宿主。jev感染的患者多数呈隐性感染,即使治愈后也会留有不可逆的脑部炎症和严重的神经系统损伤。jev感染猪一般呈散发、隐性感染,可引起怀孕母猪流产,公猪的急性睾丸炎或不育等,给养猪业带来了较大的经济损失。jev入侵细胞是诱导乙型脑炎发生的先决条件,入侵机制的阐明将为乙型脑炎的防治和治疗提供更多途径。因此,控制乙型脑炎流行不仅可以减少养猪业的损失,而且对人类的健康具有重要意义。
近年来,对jev侵染细胞的机制研究不断在深入。目前发现jev能感染猪、小鼠和人源的多种类型细胞,如pk-15细胞、u251细胞、mdscs细胞、vero细胞、neuro2a细胞和hek293细胞等,过程主要分为病毒与宿主细胞表面受体识别并结合、经由内吞途径进入宿主细胞以及脱衣壳释放核酸三步,但是至今对jev侵染宿主细胞的途径还不太清楚。因此,开展jev与宿主互作的研究有助于更加深入地了解jev的感染机制,有助于研制新的抗病毒药物或开展基因编辑抗病育种工作。
目前,多种技术可用于开展病毒与宿主互作机制的研究,新近发展的基于全基因组crispr/cas9文库敲除策略是筛选抗病基因的有力途径之一。crispr/cas9技术凭借着成本低廉,操作方便,效率高等优点,成为了基因功能研究的有力帮手,是继“锌指核酸内切酶(zfn)”、“类转录激活因子效应物核酸酶(talen)”之后出现的“第三代基因组定点编辑技术”。与前两代技术相比,crispr/cas9技术最大的突破是不仅可以对单个基因进行编辑,更重要的是可以同时对多个基因进行编辑,这也为全基因组筛选提供了有效的方法。该技术主要流程是设计和构建靶向该物种全基因组的sgrna文库,包装慢病毒后感染cas9稳定表达细胞株,再利用流式细胞术富集阳性突变体细胞库,随后采用病毒分别感染野生型和突变体细胞库,富集存活的细胞进行高通量测序鉴定筛选到的sgrna,映射对应的宿主因子。
本发明以emc3基因为研究对象,通过crispr/cas9技术构建了emc3基因编辑细胞株,并结合一系列分子和病毒学实验,证明在猪源肾细胞(pk-15)中敲除emc3基因,缺失emc3蛋白表达可显著干扰jev复制。经文献调研发现,目前尚无关于emc3基因参与介导jev复制的研究报道。因此,本发明为抗jev感染药物研发和开展猪的抗病育种研究提供了新的素材。
技术实现要素:
本发明的目的在于提供emc3基因的新应用,缺失表达emc3蛋白,可显著降低jev在宿主细胞中的复制能力,该基因可作为抗乙型脑炎的基因编辑靶点或用于抗乙型脑炎病毒药物的开发。
本发明的另一目的在于提供定点敲除emc3基因的方法,利用crispr/cas9慢病毒和流式分选技术,对emc3基因的蛋白编码序列进行移码突变,使emc3蛋白表达缺失,从而制备emc3基因敲除细胞株。
为了实现上述目的,本发明采取以下技术措施:
emc3基因的应用,具体是,采用基因工程技术(比如zfn,talen和crispr/cas9技术等)缺失表达emc3蛋白,从而抑制jev在宿主细胞中复制。
优选地,采用crispr/cas9系统敲除emc3基因,采用的sgrna靶向序列为emc3基因第一外显子。
emc3基因作为靶点,在制备防治动物感染流行性乙型脑炎病毒的药物中的应用。
emc3基因作为靶点,在制备对流行性乙型脑炎具有抗性的基因编辑细胞或动物模型中的应用。
利用crispr/cas9技术,构建emc3基因敲除细胞株,其构建方法如下:
(1)利用软件设计靶向emc3基因蛋白编码区的特异sgrna,将sgrna片段克隆到载体上,构建带有gfp筛选标记的目的sgrna表达载体;
(2)通过慢病毒包装及感染的方法将目的sgrna表达质粒导入到稳定表达cas9蛋白的细胞中,利用流式细胞仪分选gfp阳性细胞,再通过ta克隆检测dna序列和westernblot技术检测emc3蛋白的表达情况,挑选出emc3蛋白缺失的基因敲除细胞株。
优选地,所述sgrna的核苷酸序列为5′-cgattggcaggaccacccagagg-3′,其中包含的pam序列为agg。
通过对猪、人和小鼠的emc3基因的核苷酸和氨基酸序列进行多序列比对,发现不同物种来源的emc3基因序列高度保守,因此基因编辑不同物种来源emc3基因后,均能抑制jev复制。
与现有技术相比,本发明具有以下优点:
(1)本发明综合采用多种方法评估靶向编辑emc3基因对jev在宿主细胞中复制的影响,所采用的主要技术手段有:利用绝对定量和空斑实验检测不同时间点emc3基因敲除细胞株对jev编码c基因的拷贝数和病毒滴度的影响;利用免疫荧光技术检测敲除emc3基因对jev编码ns3基因的表达影响;利用rtca实时无标记细胞分析技术评估emc3基因敲除细胞对jev诱导细胞死亡的抵抗力。以多种不同的实验技术由多个层面分别验证敲除emc3基因对jev在pk-15细胞中复制的影响,提高了结果的准确性。
(2)流行性乙型脑炎是由jev引起的一种人畜共患传染病。猪感染后表现为高热、流产、产死胎及睾丸炎。尽管乙型脑炎疫苗是预防流行性乙型脑炎的有效措施,但由于jev感染机制尚不清楚,目前尚无有效的治疗药物,主要是缺乏有效分子靶点。本发明通过分子和病毒学实验研究发现,利用基因编辑技术定点敲除人、小鼠和猪高度保守的emc3基因,使emc3蛋白表达缺失,可显著抑制jev复制,同时证明emc3基因是参与jev复制的关键宿主因子。因此,本发明为抗jev药物开发、基因编辑细胞与动物模型制备提供了新的靶点。
(3)本发明还提供了一种高效构建emc3基因敲除细胞模型的方法,优选crispr/cas9技术,提供了一条采用软件设计和筛选到的特异高效sgrna序列,可用于高效制备emc3蛋白缺失细胞株。本发明方法还为构建emc3基因敲除动物模型提供了技术参考与载体材料。
附图说明
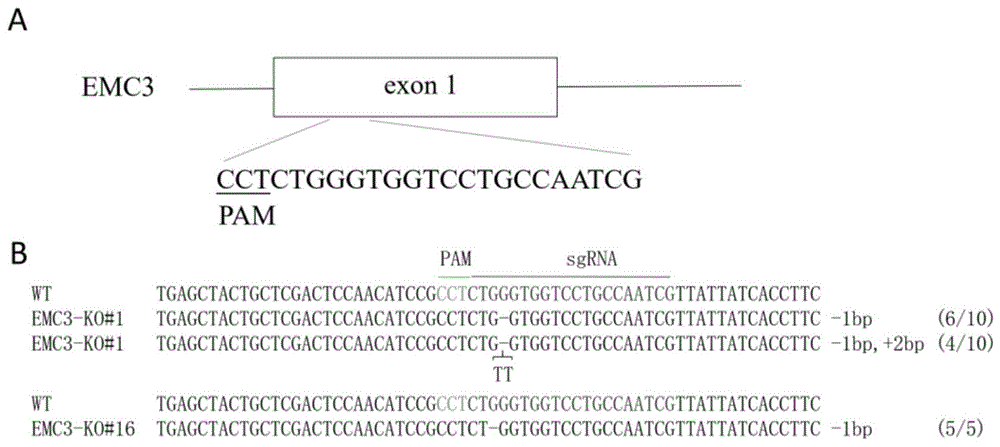
图1.采用crispr/cas9技术构建emc3基因敲除细胞株
a.利用软件设计靶向emc3基因第一外显子的sgrna示意图;b.通过ta克隆测序检测2株emc3基因编辑细胞株的基因型。bp代表碱基对,ko代表敲除,-代表删除的碱基数,+代表插入的碱基数,(n/m)代表该基因型在ta克隆测序中出现的次数。pam代表前间区序列邻近基序(protospaceradjacentmotif)。图2.利用westernblot技术检测基因敲除细胞株中emc3蛋白的表达情况β-tubulin即微管蛋白,其作为阳性内参,wt代表未进行crispr/cas9编辑的对照组细胞。
图3.利用空斑实验评价敲除宿主细胞的emc3基因对jev复制的影响
wt代表未进行crispr/cas9编辑的对照组细胞,mock代表未进行jev处理的wt细胞,hpi代表病毒感染后的时间。
图4.利用荧光定量pcr实验评价敲除宿主细胞的emc3基因对jev编码c基因的拷贝数的影响
wt代表未进行crispr/cas9编辑的对照组细胞。
图5.利用免疫荧光实验检测敲除宿主细胞emc3基因对jev编码基因ns3表达的影响
wt代表未进行crispr/cas9编辑的对照组细胞,dapi代表对细胞核进行染色,ns3代表jev编码的基因,merge代表dapi和ns3合并。
图6.利用细胞生长实验比较jev感染72h后emc3基因敲除细胞与野生型细胞的数量变化
wt代表未进行crispr/cas9编辑的对照组细胞,moi代表病毒感染复数,hpi代表病毒感染后的时间,emc3-ko(sorted)代表慢病毒感染后流式分选富集的gfp阳性细胞。
图7.利用rtca实时无标记细胞分析技术评估emc3基因敲除细胞株对jev诱导细胞死亡的抵抗力
wt代表未进行crispr/cas9编辑的对照组细胞,jevadded代表在此时间点向细胞中加入jev-rp9。
图8.猪emc3基因序列多物种保守性分析
具体实施方式
本发明所述技术方案,如未特别说明,均为本领域的常规方案,所述试剂或生物材料,如未特别说明,均已公开。
实施例1:利用基因编辑技术构建emc3基因敲除细胞株
首先,分别从ensemble数据库(www.ensembl.org)下载猪emc3基因外显子序列(登录号:enssscg00000011562.3)和猪的全基因组序列(版本号:sus_scrofa.sscrofa11.1),然后,利用sgrnacas9软件(www.biootools.com)设计靶向猪emc3基因的sgrna(表1),根据特异性评估结果选择最佳的sgrna,sgrnaid为emc3_a_47,靶点序列为“cgattggcaggaccacccagagg”,全基因组脱靶评估发现其不存在1和2个碱基错配的脱靶位点(图1a)。
表1利用软件设计和评估靶向猪emc3基因的sgrna
进一步,基于lenti-sgrna-egfp慢病毒载体为骨架,设计并合成sgrna引物,如下emc3-sgrna-f:5′-caccgcgattggcaggaccacccag-3′,emc3-sgrna-r:5′-aaacctgggtggtcctgccaatcgc-3′,引物退火后与酶切线性化的lenti-sgrna-egfp载体连接构建目的质粒。具体反应体系和条件如下:
各取5μl的emc3-sgrna-f(10μm)与emc3-sgrna-r(10μm)涡旋混匀,pcr仪上95℃,10min;65℃,1h进行变性和退火,再与bbsi(neb)酶切和纯化回收的线性化lenti-sgrna-egfp载体混匀,16℃静置连接30min。
将连接产物通过热激法转化进入感受态大肠杆菌dh5a,然后涂布在amp+抗性的lb固体培养皿中,置于37℃培养箱中过夜培养,挑选单克隆菌落扩大培养后送公司测序,测序引物为u6-f:5′-actatcatatgcttaccgtaac-3′。取测序鉴定成功的菌液扩大培养,利用omega去内毒素质粒提取试剂盒提取质粒,命名为lenti-sgrna-emc3。然后,包装lenti-sgrna-emc3慢病毒,感染pk-15细胞构建emc3基因敲除细胞株,具体实验流程如下:
前一天,接种hek293t细胞于10cm2培养皿中,待汇集度70%-90%时进行慢病毒包装。首先,取总量24μg质(pspax2:pmd2.g:lenti-sgrna-emc3=1:2:3)加入500μl的jetprimebuffer中,涡旋混匀,再加入40μl的jetprimetransfectionregent,振荡混匀,静置10min。然后,将上述溶液加入提前换液为5ml2%fbs培养基的10cm2培养皿中,放回37℃5%co2培养箱中培养。第6h换液为10ml2%fbs培养基,第24h补加10ml相同的培养基,继续培养至第60h收取上清液,以4℃30000rpm/min离心3h,倒尽上清,取200μl预冷的pbs重悬慢病毒沉淀,4℃过夜弥散后于-80℃冻存。
取靶向emc3基因的sgrna慢病毒感染稳定表达cas9的pk-15细胞株,培养至48h,利用流式细胞仪分选gfp阳性细胞,同时分选单个细胞至96孔培养板挑选单克隆细胞株。从ensemble数据库下载猪emc3基因序列,设计检测sgrna靶标区域的pcr引物,emc3-pcr-f:5′-ccacttacccgccaagaagt-3′和emc3-pcr-r:5′-gatgagaaaccccggcaaac-3′。利用tiangen血液/组织/细胞dna提取试剂盒提取的单克隆细胞dna为模板进行pcr扩增,具体pcr反应体系和条件如下:
pcr反应体系:
pcr反应条件:
pcr扩增完成后,纯化pcr产物与pmd19-t载体连接,转化涂板培养,每次挑取5个单克隆菌落扩大培养,送公司测序鉴定基因型。对比野生型细胞株基因序列,发现emc3-ko#1,emc3-ko#16两个细胞株emc3基因发生移码突变。如图1所示,emc3-ko#1有2种形式的碱基序列变化,为缺失1个碱基,同时缺失1个和插入2个碱基;emc3-ko#16仅有一种形式的碱基变化,为缺失1个碱基。
进一步,通过westernblot技术检测这2个细胞株的emc3蛋白表达情况,具体实验流程如下:
接种单克隆细胞于六孔培养板中,设置稳定表达cas9的pk-15野生型细胞作为对照组,待汇集度达到90%左右,每孔加入1mmpmsf和100μlripa裂解液,冰浴30min裂解细胞,4℃13000rpm离心20min,收集上清液,用bca蛋白定量试剂盒测定浓度。取40μg变性后的蛋白样品进行聚丙烯酰胺凝胶电泳,根据emc3蛋白分子量大小切割目的条带,同时切割β-tubulin蛋白电泳条带作为阳性内参,利用湿转膜法将两组蛋白分别同时转移至pvdf膜,用脱脂奶粉封闭后,一抗4℃孵育过夜,孵二抗后显影。如图2所示,3株细胞β-tubulin蛋白表达良好,emc3-ko#1,emc3-ko#16两株细胞中emc3蛋白完全不表达,而野生型细胞中emc3蛋白表达良好,结果表明emc3基因敲除细胞株构建成功。
实施例2:绝对定量和空斑实验发现敲除emc3基因可显著抑制jev在宿主细胞中的复制能力
为了检测敲除emc3基因是否能够抑制jev在pk-15细胞中的复制,利用绝对定量和空斑实验检测emc3-ko#1和emc3-ko#16两株单克隆细胞株对jev复制的影响,具体实验流程如下:
首先,同时接种多组emc3-ko#1和emc3-ko#16细胞,并设置稳定表达cas9的pk-15野生型细胞作为对照组。待汇集度达到50%左右,每孔按照moi=1加入对应体积的jev-rp9野生型病毒,摇匀后放回细胞培养箱培养,第2h换为2%fbs培养基继续培养,于2h、12h、24h、36h和48h等不同时间点,分别收集培养板置于-80℃反复冻融2次,取含病毒的培养液进行绝对定量和空斑实验。
空斑实验前一天,接种bhk细胞于十二孔培养板,待汇集度达到40%左右进行接毒。首先,将病毒培养液按照倍比稀释法用dmem稀释,得到不同稀释度的病毒悬液;然后,取各取500μl感染bhk细胞,每个稀释度设置三组重复。接毒第2h换液为1ml含2%fbs、1%双抗和50%低熔点琼脂糖的dmem(2×)培养基,室温静置,待培养基完全凝固后,放回培养箱继续培养待产生明显的空斑时进行10%中性甲醛固定和结晶紫染色,按照病毒滴度=空斑数/病毒悬液体积/稀释度公式计算病毒滴度(pfu/ml)。如图3所示,相同稀释度的情况下,与野生型对照组比,第2h各组产生的空斑数目基本相似,说明emc3基因对jev的入侵无显著影响;第12h、24h和36h,emc3-ko#1和emc3-ko#16细胞产生的空斑明显减少,病毒滴度明显降低,统计学检验发现emc3-ko#1和emc3-ko#16组与对照组存在显著差异,第12h差异最显著。
每个处理组各取200μl含病毒的培养液进行绝对定量实验,检测jev的拷贝数。首先,利用takara试剂盒提取病毒rna,然后反转录得到cdna,反转录具体方法如下:
1.去除基因组dna反应
反应条件为:42℃,2min;4℃,2min。
2.反转录反应(sybrgreenqpcr法)
反应条件为:37℃,15min;85℃,5s;4℃,2min。
以上述cdna为模板,按照syrbgreenqpcr法进行荧光定量pcr扩增。首先,针对jev编码的c基因设计合成特异的定量pcr引物,如下:jev-c-f:5′-gagcttgttggacggcagag-3′和jev-c-r:5′-cacggcgtcgatgagtgttc-3′;然后,以倍比稀释的编码jev-c基因的质粒为模板进行荧光定量pcr扩增,得到ct值与拷贝数的相互关系,绘制标准曲线;再对实验样本进行扩增,每组设置三个重复。荧光定量pcr具体反应体系和条件如下:
反应体系:
反应条件为:
针对荧光定量pcr得到的ct值,根据标准曲线计算对应的病毒拷贝数。如图4所示,相同稀释度的情况下,与野生型对照组比,第2h各组的病毒拷贝数基本相似,说明emc3基因对jev的入侵无显著影响;第12h,24h,36h和48h,emc3-ko#1和emc3-ko#16的病毒拷贝数明显降低,统计学检验发现emc3-ko#1和emc3-ko#16组与对照组存在显著差异,第12h差异最显著。
实施例3:利用免疫荧光实验发现敲除emc3基因可显著抑制jev编码蛋白在宿主细胞中表达
进一步,利用免疫荧光实验检测jev感染emc3-ko#1和emc3-ko#16细胞第12h时jev编码基因ns3的表达情况。具体实验流程如下:
接种emc3-ko#1和emc3-ko#16细胞,设置稳定表达cas9的pk-15野生型细胞作为对照,待汇集度达到90%左右,每孔按照moi=1加入对应体积的jev-rp9野生型病毒,摇匀后放回细胞培养箱培养,第2h换为2%fbs培养基继续培养至第12h。取第12h的细胞用多聚甲醛pfa固定,经0.3%tritonx-100处理后加入封闭液室温封闭1h,ns3(jev)一抗4℃孵育过夜,二抗室温避光孵育2h后dapi染色即进行荧光成像。结果如图5所示,不接种jev的野生型细胞中无ns3蛋白表达,接种jev的野生型细胞中ns3蛋白大量表达,接种jev的emc3-ko#1和emc3-ko#16细胞中ns3蛋白完全不表达。总之,免疫荧光实验证明敲除emc3基因显著影响jev编码基因ns3在pk-15细胞中的表达。
实施例4:jev感染emc3基因敲除细胞株致细胞病变效应观察
为了直接观察敲除emc3基因后,细胞受jev感染所导致的病变效应和细胞存活状态,利用rtca(realtimecellularanalysis)和倒置显微镜细胞成像技术同时进行检测。
利用倒置显微镜细胞成像技术监测了接种不同moi的jev对细胞生长产生的影响。首先,接种流式分选的gfp阳性细胞群即emc3-ko(sorted)细胞,设置稳定表达cas9的pk-15野生型细胞作为对照,待汇集度达到40%左右,按照不同moi加入对应体积的jev-rp9野生型病毒,摇匀后放回细胞培养箱培养,第2h换为2%fbs培养基继续培养至第72h,观察细胞存活状态,其中moi设置0.03和1两个梯度。如图6所示,不同moi感染时,野生型处理组细胞存活率极低或者几乎无存活,而emc3-ko(sorted)组,细胞大量存活,几乎无死亡。
利用rtca(rochertcadp)实时监测jev感染后细胞的存活状态。接种大致相同数量的emc3-ko#1和emc3-ko#16细胞,设置稳定表达cas9的pk-15野生型细胞作为对照,按照moi=1接种对应体积的jev,置于rtca仪上每隔15min读取一次存活细胞数量,至第60h得出细胞生长曲线。wt代表稳定表达cas9的pk-15野生型细胞,mock代表不接种jev。结果如图7所示,wt-jev组在接种jev第20h左右细胞数量呈现极具减少,然而除wt-jev组外,其它处理组细胞生长状况均表现为良好。两组实验结果表明,敲除emc3基因并不影响细胞正常生长,并且能够抵抗因jev复制所导致的细胞死亡。
实施例5:利用生物信息学策略分析emc3基因序列的物种保守性
从ncbi核酸数据库(https://www.ncbi.nlm.nih.gov/),分别下载来自猪(登录号:xm_003358517.4),小鼠(登录号:nm_175101.3)和人(登录号:nm_018447.3)的emc3基因的mrna序列。将mrna序列输入ncbi的orf在线软件(https://www.ncbi.nlm.nih.gov/orffinder/),可提取其cds序列,然后再利用多序列比对程序clustalw(https://www.genome.jp/tools-bin/clustalw)进行比对,结果如图8可见,猪和人emc基因的cds序列比对相似性得分为94.2748、猪和小鼠的为91.2214,而人和小鼠的为90.8397。表明猪和人的emc基因相似性相对高。将cds序列利用orf软件翻译为蛋白编码氨基酸序列,同样利用clustalw程序进行多序列比对分析,结果发现emc3基因蛋白编码氨基酸序列长度均为261个氨基酸,猪和人的emc蛋白序列比对相似性为99.6169,猪和小鼠的为99.2337,而小鼠和人的为99.6169,表明emc3基因的蛋白编码氨基酸序列非常保守,仅存在一个氨基酸的差异。推测emc3基因在猪、人和小鼠中功能高度一致。基于此,我们预测利用基因编辑技术靶向人、小鼠和猪的emc3基因进行定点敲除,均能对jev在不同物种宿主细胞中的复制产生显著的抑制作用。
- 还没有人留言评论。精彩留言会获得点赞!