一种检测羟基多环芳烃的多通道质谱衍生试剂及其制备方法与应用与流程
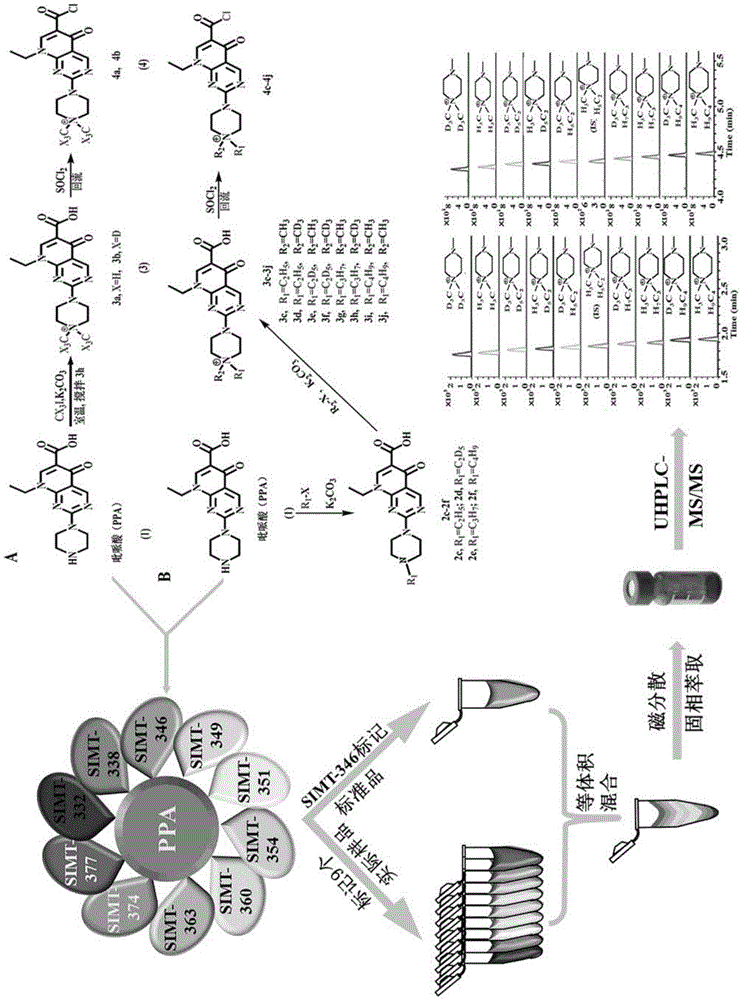
本发明属于分析化学技术领域,具体涉及一种检测羟基多环芳烃的多通道质谱衍生试剂及其制备方法与应用,尤其是涉及一种利用4’-(n,n-二烷基)-1-哌嗪基-6-碳酰氯-吡哌酸作为衍生试剂,多通道稳定同位素标记衍生-磁分散固相萃取联合超高效液相色谱三重四极杆质谱检测的多种游离羟基多环芳烃分析方法。
背景技术:
多环芳烃(polycyclicaromatichydrocarbons,pahs)暴露对人类健康构成严重威胁,例如肺癌、糖尿病、心脑血管疾病相关,同时孕妇暴露于高浓度多环芳烃经代谢会引起流产、早产、胎儿宫内生长受限等不良妊娠结局,还会影响童年时期的智力发育。羟基多环芳烃(hydroxylpolycyclicaromatichydrocarbons,oh-pahs)是评估人类多环芳烃暴露水平的生物标志物。同时测定多种不同的羟基多环芳烃在暴露研究中是必要的。在人体中,多环芳烃的代谢物大多以结合物的形式存在,且基质效应干扰严重,这对于检测痕量游离羟基多环芳烃是一个巨大的挑战。因此,建立一种快速、灵敏、准确、高通量的游离羟基多环芳烃的分析方法十分重要和必要。
中国专利(cn103175920a)公开了一种利用n,o-双三甲硅基乙酰胺作为衍生试剂,通过气相色谱-质谱联用仪对1-羟基菲,2-羟基菲,3-羟基菲,4-羟基菲,9-羟基菲,1-羟基芘,3-羟基苯并[a]芘和7-羟基苯并[a]芘分析检测方法,使用2-羟基菲-d9、1-羟基芘-d9和3-羟基苯并[a]芘-d11三个氘代化合物作为内标,衍生时间长,灵敏度低,内标化合物昂贵且不易购买;中国专利(cn106483230a)公开了一种利用丙酮和正十一醇做为对2-羟基萘,2-羟基芴,2-羟基菲,3-羟基菲,4-羟基菲和1-羟基芘进行分散液液萃取-上浮液滴固化法富集,结合高效液相色谱-荧光检测方法进行分析检测,耗时,灵敏度低,分析通量低。因此,开发一种高灵敏度、高通量、多种成分同时检测的分析方法,对人体多环芳烃暴露水平评估分析具有十分重要的意义。
技术实现要素:
本发明所要解决的技术问题是如何灵敏、准确且高通量的同时检测多种游离羟基多环芳烃,本发明提供了一种检测羟基多环芳烃的多通道质谱衍生试剂。
本发明还提供了上述检测羟基多环芳烃的多通道质谱衍生试剂的制备方法。
本发明还提供了一种利用上述多通道质谱衍生试剂检测羟基多环芳烃的的方法,本发明首次设计合成10通道稳定同位素质量标签(simt-332/338/346/349/351/354/360/363/374/377),其中simt-346标记标准品的衍生化产物作为内标,其他9种质量标签(simt-332/338/349/351/354/360/363/374/377)分别标记9个实际样品,10份溶液等体积混合后进行磁分散固相萃取联合超高效液相色谱三重四极杆质谱检测的分析方法,可以实现一次进样同时分析测定9个实际样品中的多种羟基多环芳烃。利用磁性氧化石墨烯作萃取材料,具有操作简单的优点。
本发明为了实现上述目的所采用的技术方案为:
本发明提供了一种检测羟基多环芳烃的多通道质谱衍生试剂,所述衍生试剂的结构式如下:
所述r1为ch3,r2为ch3、c2h5、c2d5、c3h7或c4h9;
所述r1为cd3,r2为cd3、c2h5、c2d5、c3h7或c4h9。
本发明还提供了一种上述多通道质谱衍生试剂的制备方法,包括以下步骤:
a.将0.5g吡哌酸溶解在20ml乙腈中,然后加入0.59gk2co3,并在室温搅拌下加入6.10mmol碘甲烷或氘代碘甲烷,将反应混合物在室温搅拌8~9h,减压抽滤,得到的固体在烘箱中干燥。获得白色固体粉末即[ch3]2/[cd3]2-ppa;
b.将0.5g吡哌酸溶解在20ml乙腈中,然后加入0.59gk2co3,并在室温搅拌下分别加入6.59mmol溴乙烷、氘代溴乙烷、1-溴丙烷或正溴丁烷,将反应混合物在室温搅拌12~14h后,在搅拌下分别加入6.10mmol碘甲烷或氘代碘甲烷和0.59gk2co3,继续搅拌22~25h,减压抽滤,得到的固体在烘箱中干燥,获得白色固体粉末即[ch3-c2h5]-ppa、[cd3-c2h5]-ppa、[ch3-c2d5]-ppa、[cd3-c2d5]-ppa、[ch3-c3h7]-ppa、[cd3-c3h7]-ppa、[ch3-c4h9]-ppa、[cd3-c4h9]-ppa;
c.将1.0ga和b中产物和25ml亚硫酰氯置于50ml反应烧瓶中,该反应烧瓶连接至具有naoh填充管的回流冷凝管。将该混合物在80℃下搅拌、回流3小时,减压蒸除氯化亚砜,冷却至室温,得橙黄色固体即为4’-(n,n-二烷基)-1-哌嗪基-6-碳酰氯-吡哌酸(simt-332/338/346/349/351/354/360/363/374/377)。
本发明还提供了一种利用上述多通道质谱衍生试剂检测羟基多环芳烃的方法,包括以下步骤:将10通道稳定同位素质量标签4’-(n,n-二烷基)-1-哌嗪基-6-碳酰氯-吡哌酸(simt-332/338/346/349/351/354/360/363/374/377)对目标物进行衍生反应:其中simt-346标记标准品的衍生化产物作为内标,其他9种质量标签(simt-332/338/349/351/354/360/363/374/377)分别标记9个实际样品,10份溶液等体积混合后使用磁性氧化石墨烯进行磁分散固相萃取,萃取液经滤膜过滤后进行液-质分析检测羟基多环芳烃。
本发明提供的检测方法具体包括以下步骤:
a.多通道稳定同位素标记衍生化过程:分别取9份10-50μl待测样品或7种标准品混合溶液,50-300μlph8.0-10的nahco3-na2co3缓冲溶液,分别加入离心管中,然后分别注入100-600μl4’-(n,n-二烷基)-1-哌嗪基-6-碳酰氯-吡哌酸乙腈溶液(simt-332/338/349/351/354/360/363/374/377)和4’-(n,n-二烷基)-1-哌嗪基-6-碳酰氯-吡哌酸(simt-346)乙腈溶液,摇匀后密封,在温度35-50℃水浴中振荡反应3-5分钟;得待测物衍生化产物溶液和内标物溶液;
b.磁分散固相萃取过程:取50-100μl待测物衍生化产物溶液和内标溶液等体积的混合溶液,加入6-10mg萃取材料,通过超声辅助将其分散形成悬浮液,然后使用硼酸盐缓冲溶液将溶液的ph调节至7.0,剧烈振荡吸附3-6分钟,然后通过外部磁铁分离混合物,最后在100-500μl解吸剂溶液中涡旋1-5分钟进行解吸操作,然后再氮气流下干燥并将固体用乙腈重新溶解,得到羟基多环芳烃衍生萃取物溶液;
c.将羟基多环芳烃衍生萃取物溶液经滤膜过滤后,利用超高效液相色谱三重四极杆串联质谱系统进行分析检测。
进一步的,所述标准品混合溶液的浓度是1.0×10-5mol/l。
本发明所使用的萃取材料为磁性氧化石墨烯,具体采用以下方法制备而成:将0.1ggo加入到100ml高纯水中,超声2h,得到1g/ml的go分散溶液,同时用氮气吹扫5.40gfecl3·6h2o和1.98gfecl2·4h2o在30ml水中的溶液,然后将上述溶液和go溶液在氮气下搅拌3h,加入28%的氨溶液调节溶液的ph值,合成磁铁矿材料,在60℃下快速搅拌3h,溶液冷却至室温,然后,用水/乙醇再次洗涤深黑色溶液三次,并在65℃的真空中干燥,得到fe3o4/go复合材料。
进一步的,所述解吸剂为甲醇,乙醇,乙腈,丙酮,甲醇(0.1%甲酸),甲醇(0.5%甲酸),甲醇(1%甲酸),甲醇(5%甲酸)或甲醇(10%甲酸);优选的,所述解吸剂为甲醇(1%甲酸);所述解析时间为2分钟。
本发明的检测目标物,游离羟基多环芳烃包括1-羟基芘、1-羟基萘、2-羟基萘、2-羟基芴、3-羟基菲、4-羟基菲和9-羟基菲中的一种或多种。
本发明所使用的超高效液相色谱三重四极杆串联质谱系统中色谱分离使用安捷伦sbc18色谱柱:2.1mm×50mm,1.8μm,进样体积为2μl,柱温30℃,采用梯度洗脱法;所述的梯度洗脱法,时间为5.5min,流速为0.2ml/min,流动相a为10%甲醇水溶液含有0.1%甲酸,流动相b为甲醇含有0.1%甲酸,0min流动相组成为35%a+65%b,1min时15%a+85%b,3.5min时2%a+98%b,4.5min时2%a+98%b,5min时0%a+100%b,5.5min时0%a+100%b。
上述的超高效液相色谱三重四极杆串联质谱系统进行分析检测时的质谱的条件为:干燥气温度300℃,流速10l/min,喷雾器气压40psi,鞘气温度300℃,流速11l/min,毛细管电压3.5kv。
本发明提供了一种同时高通量检测多个生物样品中多种游离羟基多环芳烃的分析方法,所述的羟基多环芳烃在温和条件下与衍生试剂4’-(n,n-二烷基)-1-哌嗪基-6-碳酰氯-吡哌酸发生衍生反应,得到的衍生化产物通过磁性氧化石墨烯进行磁分散固相萃取,得到的衍生萃取物溶液经滤膜过滤后,利用超高效液相色谱三重四极杆串联质谱系统进行分析检测。
本发明流动相中各分数均指体积分数。
所述的目标分析物结构式如下所示:
本发明利用稳定同位素衍生技术联合超高效液相色谱三重四极杆质谱法,对羟基多环芳烃的检测能够带来显著的质谱增敏效果。首次使用含有一个分子内永久正电荷的衍生化试剂4’-(n,n-二烷基)-1-哌嗪基-6-碳酰氯-吡哌酸来衍生羟基多环芳烃。因此,对羟基多环芳烃通过衍生化反应得到的衍生物进行检测时,衍生产物母离子能够产生特异性的含有吡哌酸结构的报告离子,带来显著的质谱增敏检测效果。通过使用多通道质量标签去标记待测样品,可以实现单针进样同时检测9个实际样品中的多种羟基多环芳烃,该方法能够节省大量时间,实现高通量分析。通过内标法定量,该分析方法具有良好的灵敏度、选择性和准确度,并且得到良好的回收率结果。7种羟基多环芳烃所含有的活性酚羟基,能够和衍生试剂4’-(n,n-二烷基)-1-哌嗪基-6-碳酰氯-吡哌酸的碳酰氯活性反应基团迅速反应。本发明克服了常规方法的反应时间长,灵敏度低,基质干扰严重,分析通量低等问题,大大提高了液-质检测的分析通量、灵敏度、选择性和准确度。本发明为人类尿液和血浆中多种羟基多环芳烃的同时检测提供了一种高效、可靠的技术手段。以1-羟基芘为例,4’-(n,n-二烷基)-1-哌嗪基-6-碳酰氯-吡哌酸(simt-332)为衍生试剂的衍生化反应过程如下所示:
本发明的优点及有益效果:
1.本发明首次设计合成10通道稳定同位素质量标签4’-(n,n-二烷基)-1-哌嗪基-6-碳酰氯-吡哌酸,用于羟基多环芳烃的稳定同位素标记衍生,其中simt-346标记的标准品作为内标,其余9种质量标签分别标记9个实际样品。实现了一次进样同时检测9个实际样品的高通量分析。衍生化反应条件温和、快速,衍生产物稳定,分析通量高,该技术显著提高了分析方法的分析通量、色谱分离度、质谱检测灵敏度。
2.本发明所提供的多通道稳定同位素标记衍生-磁分散固相萃取的前处理技术,联合超高效液相色谱三重四极杆质谱多反应监测模式,具有高通量、高灵敏、高准确、操作简便等优点。
3.本发明的检测分析方法成功应用于人类尿液和血浆中多种游离的羟基多环芳烃的检测,方法适用性良好。
附图说明
图1为多通路稳定同位素标记衍生化结合磁分散固相萃取和超高相液相色谱策略原理示意图。
图2为实施例1中10种稳定同位素质量标签分别与7种羟基多环芳烃衍生物的质谱检测分离图,(a):2-羟基萘衍生物,(b):1-羟基萘衍生物,(c):2-羟基芴衍生物,(d):3-羟基菲衍生物,(e):9-羟基菲衍生物,(f):4-羟基菲衍生物,(g):1-羟基芘衍生物。
图3为实施例1中simt-332与1-羟基芘衍生物的质谱裂解机理示意图。
图4为实施例2尿液中羟基多环芳烃的衍生物质谱检测分离图谱,(a):2-羟基萘衍生物,(b):1-羟基萘衍生物,(c):2-羟基芴衍生物,(d):3-羟基菲衍生物,(e):9-羟基菲衍生物,(f):4-羟基菲衍生物,(g):1-羟基芘衍生物。
具体实施方式
下面通过实施例对本发明进行进一步的阐述,下述说明仅为了解释本发明,并不对其内容进行限定。
本发明中衍生化萃取物溶液经0.22μm滤膜过滤后利用超高效液相色谱三重四极杆串联质谱系统进行分析检测,图1为本发明方法概览图。
本发明所使用的衍生化试剂4’-(n,n-二烷基)-1-哌嗪基-6-碳酰氯-吡哌酸合成方法如下:
a.将0.5g吡哌酸(1.65mmol),20ml乙腈加入50ml单口烧瓶中。然后加入0.59gk2co3(4.29mmol),并在室温搅拌下加入6.10mmol碘甲烷或氘代碘甲烷。将反应混合物在室温反应8~9h,减压抽滤,得到的固体在烘箱中干燥。获得白色固体粉末即[ch3]2/[cd3]2-ppa,产率99.37%/99.42%。
b.将0.5g吡哌酸(1.65mmol),20ml乙腈加入50ml单口烧瓶中。然后加入0.59gk2co3(4.29mmol),并在室温搅拌下加入6.59mmol溴乙烷、氘代溴乙烷、1-溴丙烷或正溴丁烷。室温反应12h后,在搅拌下分别加入6.10mmol碘甲烷或氘代碘甲烷和0.59gk2co3(4.29mmol)。继续搅拌22h,减压抽滤,得到的固体在烘箱中干燥。获得白色固体粉末即[ch3-c2h5]-ppa、[cd3-c2h5]-ppa、[ch3-c2d5]-ppa、[cd3-c2d5]-ppa、[ch3-c3h7]-ppa、[cd3-c3h7]-ppa、[ch3-c4h9]-ppa、[cd3-c4h9]-ppa,产率都大于92%。
c.将约1.0ga和b中产物4’-(n,n-二烷基)-1-哌嗪基-6-碳酰氯-吡哌酸和25ml亚硫酰氯置于50ml反应烧瓶中,该反应烧瓶连接至具有naoh填充管的回流冷凝管。将该混合物在80℃下搅拌、回流3小时。减压蒸除氯化亚砜,冷却至室温,得橙黄色固体即为4’-(n,n-二烷基)-1-哌嗪基-6-碳酰氯-吡哌酸(simt-332/338/346/349/351/354/360/363/374/377)。
本发明所使用的萃取材料磁性氧化石墨烯采用以下方法制备而成:将0.1ggo加入到100ml高纯水中,超声2h,得到1g/ml的go分散溶液。同时用氮气吹扫5.40gfecl3·6h2o和1.98gfecl2·4h2o在水(30ml)中的溶液。然后将上述溶液和go溶液在氮气下搅拌3h,加入28%的氨溶液调节溶液的ph值,合成磁铁矿材料。在60℃下快速搅拌3h,溶液冷却至室温。然后,用水/乙醇再次洗涤深黑色溶液三次,并在65℃的真空中干燥。得到fe3o4/go复合材料。
实施例1
7种羟基多环芳烃的色谱分离和质谱定性定量分析:
7种羟基多环芳烃标准品(购自sigma、j&k和aladdin试剂公司)用乙腈配制得到浓度为1.0×10-2mol/l的羟基多环芳烃标准品溶液。我们将无分析物的尿液以高速离心,除去杂质,将不含分析物的血浆加入乙腈(1:3,v/v),离心5分钟(12000rpm),分离上清液使用,处理后的无分析尿液和血浆按相同比例混合,作为空白基质。从每个单标储备液中各取0.2ml使用空白基质定容至10ml得到羟基多环芳烃的混标储备液。4’-(n,n-二烷基)-1-哌嗪基-6-碳酰氯-吡哌酸溶于乙腈得到1.0×10-3mol/l衍生试剂溶液;分别取9份20μl标准品溶液置于1.5ml的离心管中,另取20μl标准品混合溶液(1.0×10-5mol/l)置于1.5ml的离心管中,分别加入100μlnahco3-na2co3(ph9.2)缓冲溶液,涡旋混匀器震荡15s,分别加入200μl其他9种simts(simt-332/338/349/351/354/360/363/374/377)溶液,simt-346溶液,封口,在40℃反应5分钟。然后将simt-346标记的标准品衍生化产物溶液(内标溶液)和其他9种衍生试剂标记的标准品衍生化产物溶液在1.0ml试管中等体积混合(衍生产物混合溶液),随后用于mdspe程序。
将8mg吸附剂(fe3o4/go纳米复合材料)通过超声辅助分散在衍生产物混合溶液中形成均匀的悬浮液。然后通过使用硼酸盐缓冲液将溶液的ph调节至7.0。剧烈振荡5分钟达到吸附平衡,然后通过外部磁铁分离混合物。在200μl含1%甲酸的甲醇溶液(作为解吸剂,当该处解吸剂改用甲醇,乙醇,乙腈,丙酮,甲醇(0.1%甲酸),甲醇(0.5%甲酸),甲醇(5%甲酸)或甲醇(10%甲酸)时,分析物萃取效率处于甲醇(1%甲酸)做解吸剂时的37-88%之间)中涡旋2分钟进行解吸操作。收集洗脱液并在温和的氮气流下干燥,并将固体物质重新溶解然后进行uhplc-ms/ms分析。时间为5.5min,流速为0.2ml/min,流动相a为10%甲醇水溶液含有0.1%甲酸,流动相b为甲醇含有0.1%甲酸,0min流动相组成为35%a+65%b,1min时15%a+85%b,3.5min时2%a+98%b,4.5min时2%a+98%b,5min时0%a+100%b,5.5min时0%a+100%b;质谱的条件为:干燥气温度300℃,流速10l/min,喷雾器气压40psi,鞘气温度300℃,流速11l/min,毛细管电压3.5kv。
按照上述梯度洗脱程序能得到较好的分离度,图2为10种稳定同位素质量标签分别与7种羟基多环芳烃衍生物的质谱检测分离图,(a):2-羟基萘衍生物,(b):1-羟基萘衍生物,(c):2-羟基芴衍生物,(d):3-羟基菲衍生物,(e):9-羟基菲衍生物,(f):4-羟基菲衍生物,(g):1-羟基芘衍生物,70种羟基多环芳烃衍生物之间分离度良好。质谱分析实验结果表明,70种羟基多环芳烃衍生物多反应监测模式(mrm)下产生相同的主要子离子m/z332.2、m/z338.2、m/z346.2、m/z349.2、m/z351.2、m/z353.2、m/z360.2、m/z363.2、m/z374.2和m/z377.2用作定量子离子。以simt-332衍生1-羟基芘的衍生物为例,图3为simt-332与1-羟基芘衍生物的质谱裂解机理示意图,母离子为[m]+m/z532.2能够产生一个特异性子离子m/z332.2。对mrm模式的参数进行了优化,70种羟基多环芳烃衍生物的定量离子对、最优化碎裂电压(v)和碰撞能(ev),以及分析方法的相关系数、检出限、定量限见表1。
表1
实施例2
尿液中游离羟基多环芳烃的检测分析,包括以下操作步骤:
取500μl吸烟者或非吸烟者的尿液高速离心以除去杂质,然后取9份50μl尿液样本置于1.5ml离心管中,另取50μl标准品混合溶液(1.0×10-5mol/l)置于1.5ml的离心管中,分别加入250μlnahco3-na2co3(ph9.8)缓冲溶液,涡旋混匀器震荡15s,分别加入550μl其他9种simts(simt-332/338/349/351/354/360/363/374/377)溶液,simt-346溶液,封口,在40℃反应4.5分钟。然后将simt-346标记的标准品衍生化产物溶液(内标溶液)和其他9种衍生试剂标记的尿液样品衍生化产物溶液在1.0ml试管中等体积混合(衍生产物混合溶液),随后用于mdspe程序。
将7mg吸附剂(fe3o4/go纳米复合材料)通过超声辅助分散在衍生产物混合溶液中形成均匀的悬浮液。然后通过使用硼酸盐缓冲液将溶液的ph调节至7.0。剧烈振荡5.5分钟达到吸附平衡,然后通过外部磁体分离混合物。在450μl含1%甲酸的甲醇溶液中涡旋3分钟进行解吸操作。收集洗脱液并在温和的氮气流下干燥,并将固体物质重新溶解然后进行uhplc-ms/ms分析。检测到吸烟者尿液中游离羟基多环芳烃含量(n=3)分别为:1-羟基芘0.0492ng/ml、1-羟基萘0.7644ng/ml、2-羟基萘1.3847ng/ml、2-羟基芴0.2486ng/ml、3-羟基菲0.0273ng/ml、4-羟基菲0.0135ng/ml、9-羟基菲0.1419ng/ml;非吸烟者尿液中游离羟基多环芳烃含量(n=3)分别为:1-羟基芘0.0042ng/ml、1-羟基萘0.2171ng/ml、2-羟基萘0.4069ng/ml、2-羟基芴0.0614ng/ml、3-羟基菲0.0115ng/ml、4-羟基菲0.0042ng/ml、9-羟基菲0.0077ng/ml,该样品质谱检测图见图4。
实施例3
血浆中游离羟基多环芳烃的检测,包括以下操作步骤:
取200μl吸烟者或非吸烟者的血浆,加入100μl乙腈,高速离心5分钟(12000rpm)并分离上清液以供使用。取9份20μl血浆样本置于1.5ml离心管中,另取20μl标准品混合溶液(1.0×10-5mol/l)置于1.5ml的离心管中,分别加入100μlnahco3-na2co3(ph9.0)缓冲溶液,涡旋混匀器震荡15s,分别加入150μl其他9种simts(simt-332/338/349/351/354/360/363/374/377)溶液,simt-346溶液,封口,在40℃反应6分钟。然后将simt-346标记的标准品衍生化产物溶液(内标溶液)和其他9种衍生试剂标记的血浆样品衍生化产物溶液在1.0ml试管中等体积混合(衍生产物混合溶液),随后用于mdspe程序。
将8.5mg吸附剂(fe3o4/go纳米复合材料)通过超声辅助分散在衍生产物混合溶液中形成均匀的悬浮液。然后通过使用硼酸盐缓冲液将溶液的ph调节至6.8。剧烈振荡4.7分钟达到吸附平衡,然后通过外部磁体分离混合物。在240μl含1%甲酸的甲醇溶液中涡旋1.7分钟进行解吸操作。收集洗脱液并在温和的氮气流下干燥,并将固体物质重新溶解然后进行uhplc-ms/ms分析。吸烟者血浆中检测到3种游离羟基多环芳烃含量(n=3)分别为:1-羟基萘0.0084ng/ml、2-羟基萘0.0151ng/ml、2-羟基芴0.0007ng/ml;非吸烟者血浆中仅检测到2种游离羟基多环芳烃含量(n=3)分别为:1-羟基萘0.0003ng/ml、2-羟基萘0.0008ng/ml。
实施例4
沉积物中羟基多环芳烃的检测,包括以下操作步骤:
参考文献(中山大学学报(自然科学版),2015,02:77-82),取9份河流沉积物样品2.0g经冷冻干燥,研磨过0.125mm的金属筛用甲醇进行加速溶剂萃取,再将萃取出的样品旋转蒸发浓缩后,以正己烷再次溶解并过中性氧化铝柱;以含2%醋酸的二氯甲烷作为洗脱溶剂,重复洗脱两次,每次5ml;洗脱出来的样品用氮气吹干,加入20μl乙腈备用,另取20μl标准品混合溶液(1.0×10-5mol/l)置于1.5ml的离心管中,分别加入150μlnahco3-na2co3(ph9.5)缓冲溶液,涡旋混匀器震荡15s,分别加入200μl其他9种simts(simt-332/338/349/351/354/360/363/374/377)溶液,simt-346溶液,封口,在45℃反应4.5分钟。然后将simt-346标记的标准品衍生化产物溶液(内标溶液)和其他9种衍生试剂标记的沉积物样品衍生化产物溶液在1.0ml试管中等体积混合(衍生产物混合溶液),随后用于mdspe程序。
将7.5mg吸附剂(fe3o4/go纳米复合材料)通过超声辅助分散在衍生产物混合溶液中形成均匀的悬浮液。然后通过使用硼酸盐缓冲液将溶液的ph调节至7.0。剧烈振荡5.5分钟达到吸附平衡,然后通过外部磁体分离混合物。在200μl含1%甲酸的甲醇溶液中涡旋2.3分钟进行解吸操作。收集洗脱液并在温和的氮气流下干燥,并将固体物质重新溶解然后进行uhplc-ms/ms分析。检测到某河底沉积物中7种羟基多环芳烃(n=3)分别为:1-羟基芘0.0272ng/g、1-羟基萘0.7218ng/g、2-羟基萘0.7504ng/g、2-羟基芴0.7029ng/g、3-羟基菲0.6953ng/g、4-羟基菲0.5485ng/g、9-羟基菲0.7739ng/g。
对比例1
该对比例过程与实施例2相同,不同在于衍生化反应和样品前处理过程中未进行磁分散固相萃取,衍生化试剂与衍生条件采用中国专利(cn103185762a)公开的n,o-双三甲硅基乙酰胺作为衍生试剂。
对比例2
该对比例过程与实施例2相同,不同在于衍生化反应和样品前处理过程,衍生化试剂与衍生条件采用文献(journalofchromatographya,2015,1379:51-55)公开的丹磺酰氯作为衍生试剂和液-液萃取。
对比例3
该对比例操作步骤与实施例2相同,不同在于样品前处理过程中,采用中国专利(cn106483230a)公开的未使用衍生试剂衍生和未进行磁分散固相萃取。
对比例4
该对比例过程与实施例2相同,不同在于样品前处理过程中,采用文献(分析测试学报,2019,38:270–276)公开的未使用衍生试剂衍生和未进行磁分散固相萃取。
下表2为实施例2同对比例1-4的试验结果对比。
表2
从表2中可以看出,与相关报道相比,本发明利用4’-(n,n-二烷基)-1-哌嗪基-6-碳酰氯-吡哌酸对羟基多环芳烃进行衍生化标记,衍生条件温和、快速,灵敏度高。本方法与对比例1相比,本发明省时,反应条件温和,灵敏度更高;与对比例2相比,检出限相似,但是本发明分析通量是它的9倍;与对比例3和对比例4相比,不仅分析通量高9倍,检出限高2-6个数量级。另外,本发明回收率结果良好,对分析方法的准确度提供了保障。
为了验证所建立的分析方法的适用性,对精密度、准确度(回收率)和基质效应都进行了详细考察,结果见表3。
表3
由表2和表3可知,对于9个实际样品中7种羟基多环芳烃衍生物,其线性范围在2-3000pg/ml,检出限分布于0.1-0.5pg/ml之间,定量限分布于0.5-2.0pg/ml之间。结果表明,所建立的分析方法分析通量高,灵敏度高,准确度高,有效降低了基质干扰,能够很好地应用于检测上述三种实际样品中的多种羟基多环芳烃的含量。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受实施例的限制,其它任何未背离本发明的精神实质与原理下所做的改变、修饰、组合、替代、简化均应为等效替换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!