一种1,3-N-乙基加洛糖胺的合成方法与流程
本发明涉及一种有机化学合成方法,具体地说是一种以1-n-乙基加洛糖胺合成1,3-n-乙基加洛糖胺的方法。
背景技术:
硫酸依替米星,结构式如下:
化学名称:o-2-氨基-2,3,4,6-四脱氧-6-氨基-α-d-赤型-己吡喃糖基-(1→4)-o-[3-脱氧-4-c-甲基-3(甲氨基)-β-l-阿拉伯吡喃糖基-(1→6)]-2-脱氧-n-乙基-l-链霉胺硫酸盐分子式(c21h43n5o7)2·5h2so4
分子量:1445.58
硫酸依替米星是一种庆大霉素c1a的衍生物,目前有注射剂产品上市。
其作用机理是抑制敏感菌的蛋白质合成,具有高效、安全、广谱、交叉耐药性较少等特点。
临床研究显示本品对以下感染有较好的疗效:呼吸道感染:如急性支气管炎、慢性支气管炎急性发作、社区肺部感染等;肾脏和泌尿生殖系统感染:如急性肾盂肾炎、膀胱炎、慢性肾盂肾炎或慢性膀胱炎急性发作等;皮肤软组织和其它感染:如皮肤及软组织感染,外伤、创伤和手术产后的感染及其他敏感菌感染。药品在临床使用中产生的不良反应除了与药品本身的药理活性有关外,与药品中存在的杂质也有很大的关系。
本发明经过研究发现,1,3-n-乙基加洛糖胺是硫酸依替米星原料药以及制剂中的主要杂质之一,其结构式如下:
根据本发明的研究,其为硫酸依替米星的降解杂质,在硫酸依替米星存放过程中产生,但因为含量低而难以发现,本发明为解决硫酸依替米星原料存放中的质量保证,对1,3-n-乙基加洛糖胺进行了合成并对其含量测定方法进行了研究。
为此,本发明提供一种硫酸依替米星原料药及其制剂的杂质控制方法,特别是提供一种1,3-n-乙基加洛糖胺的含量测定方法,为达到此目标,本发明进一步提供作为杂质对照品的1,3-n-乙基加洛糖胺纯品的制备方法。本发明对于提高药品质量,提高临床用药的安全性具有重大的意义。
技术实现要素:
本发明的目的在于提供一种1-n-乙基加洛糖胺的制备方法。
本发明所述的制备方法,包括以下步骤:
(1)依替米星加入双氧水降解;
(2)降解液上硅胶柱分离,分离出1-n-乙基加洛糖胺(化合物1);
(3)在1-n-乙基加洛糖胺组分中加入三乙胺,乙酸酐反应得到乙酰化产物(化合物2)
(4)产物中加入二氯甲烷,乙醛,反应溶液在0~10℃下搅拌反应,随后加入加硼氢化钾,反应0.2-1h,再加硼酸缓冲液调ph值为7-11,继续反应得到乙基胺化物(化合物4),
(5)反应结束,反应液浓缩;加入10-20%氢氧化钠搅拌,反应结束,反应液过滤得到滤液,浓缩得到1,3-n-乙基加洛糖胺粗品(化合物5);
(6)滤液除盐,分离后得到目标产物,再经重结晶得到纯品1,3-n-乙基加洛糖胺。
其中,步骤(1)加入双氧水的比例为1:1~20:1,优选15:1~18:1;
其中,步骤(2)所述硅胶柱为干法装柱进行分离,用氯仿:甲醇:氨水洗脱液洗脱,薄层色谱判断成分,浓缩1-n-乙基加洛糖胺富集物,浓缩后得到含有1-n-乙基加洛糖胺的组分;
其中,步骤(3)中,乙酸酐与1-n-乙基加洛糖胺的摩尔比1:1~10:1;加热温度在80~100℃回流1-3h。优选的,乙酸酐与1-n-乙基加洛糖胺的摩尔比4:1~7:1;加热温度在90℃回流2h;
其中,步骤(4)中加入量二氯甲烷,乙醛,硼氢化钾的量与5,2′,4′-三乙酯-1-n-乙基加洛糖胺的摩尔比分别为20:1~30:1、1:1~4:1、5:1~8:1,优选,30:1、1.5:1,5:1~6:1,用硼酸缓冲溶液调节溶液的ph值至7~11,其中硼酸缓冲液配制方法如下:(1.0g硼酸加去离子水3.0ml搅拌溶解,用氢氧化钠调节ph=10),加入量为:3ml(硼酸缓冲溶液调节溶液的ph值至10);
其中,步骤(5)中加入10-20%氢氧化钠后,需要加热搅拌回流,温度为105~140℃,回流20~25h;优选的,加热温度在125℃,回流24h;
其中,步骤(6)中所述除盐,方法如下:取除盐浓缩液,用超纯水稀释浓度为10mg/ml,采用制备液相进行分离,收集有效成分。所述制备液相,色谱条件如下:
色谱柱:gemininxc18(4.6mm×150mm,5μm);流动相:a相:水-氨水-冰醋酸(96:3.6:0.4),b相:甲醇,梯度洗脱(程序见tab.1);流速:0.8ml/min;柱温:30℃;进样量:10μl;elsd参数:漂移管温度:105℃;载气流速:2.6l/min;gain值:1。
tab.1流动相梯度
特别优选的,本发明的制备方法,包括以下步骤:
(1)将依替米星原料药中加入30%双氧水降解48h;
(2)降解液上硅胶柱分离,得到1-n-乙基加洛糖胺(化合物1);
(3)1-n-乙基加洛糖胺中,加入三乙胺8-16ml,滴加乙酸酐0.65-2.5ml反应;反应完成后降温至0~10℃得到乙酰化产物(化合物2);
(4)反应液中投入二氯甲烷10-25ml,然后在0~10℃条件下,滴加乙醛0.2-0.8ml,搅拌0.5-2h,加硼氢化钾0.2-0.9g,反应0.2-1h,加硼酸缓冲液1-5ml,搅拌1-3h;得到乙基胺化物(化合物4);
(5)反应液中加入10%氢氧化钠搅拌0.5-2h,,常压加热蒸馏至液温100℃,蒸出部分溶剂,在加入20%氢氧化钠溶液8-12ml,加热回流12-48h,降温至室温,真空抽滤得到得到1,3-n-乙基加洛糖胺粗品(化合物5);
(6)滤液用无盐水稀释,通入层析分离柱中上样,用纯化水洗柱,20-60%乙醇洗脱有机相,浓缩,浓缩液加氨水混匀,通入层析分离柱中上样,收集有效成分,浓缩,浓缩液通过制备液相方法进行分离,收集有效成分,浓缩。
本发明所述的1-n-乙基加洛糖胺的制备方法,如下所示:
其中的化合物1-5的中文名称:
化合物1:1-n-乙基加洛糖胺
化合物2:5,2′,4′-三乙酯-1-n-乙基加洛糖胺
化合物3:2′,4′,5″-三乙酯-1-n-乙基-3-亚乙基加洛糖胺
化合物4:2′,4′,5″-三乙酯-1-n-乙基-3-乙基加洛糖胺
化合物5:1,3-n-乙基加洛糖胺
本发明的制备方法,相对于现有的工艺而言,具有以下有益效果:
目前并没有1,3-n-乙基加洛糖胺相关合成工艺的报道,本发明作为唯一1,3-n-乙基加洛糖胺的合成工艺,其合成路线简单,操作便利,用时短,成本低,对环境污染小,通过本发明的方法制备得到的1,3-n-乙基加洛糖胺具有纯度高和收率高等特点,对于提高药品质量,提高临床用药的安全性具有重大的意义。
本发明得到的产物1,3-n-乙基加洛糖胺可以用于硫酸依替米星原料药及其制剂中的杂质含量测定,有关测定方法如下:
将供试品溶液和对照品溶液注入高效液相色谱仪,得到色谱图,根据色谱图计算依替米星原料药中杂质1,3-n-乙基加洛糖胺的残留量。
举例如下:称取1,3-n-乙基加洛糖胺6.47mg,稀释至25ml,取定量的硫酸依替米星原料药稀释至25ml。根据以上数据及峰面积可以计算出硫酸依替米星原料药中杂质1,3-n-乙基加洛糖胺的残留量。
色谱条件如下:
用十八烷基硅烷键合硅胶为填充剂(4.6mm×250mm,5μm或效能相当的色谱柱),以0.2mol/l三氟乙酸(含0.05%五氟丙酸,1.5g/l无水硫酸钠,0.8%(v/v)的50%氢氧化钠溶液,用50%氢氧化钠调节ph值至3.5)-乙腈(96:4)为流动相,柱温为35℃,流速为每分钟1.0ml,用积分脉冲安培电化学检测器检测,检测电极为金电极(推荐使用3mm直径),参比电极为ag/agcl复合电极,钛合金对电极,四波形检测电位,柱后加碱(50%氢氧化钠溶液1→25,推荐流速每分钟0.5ml)。
附图说明:
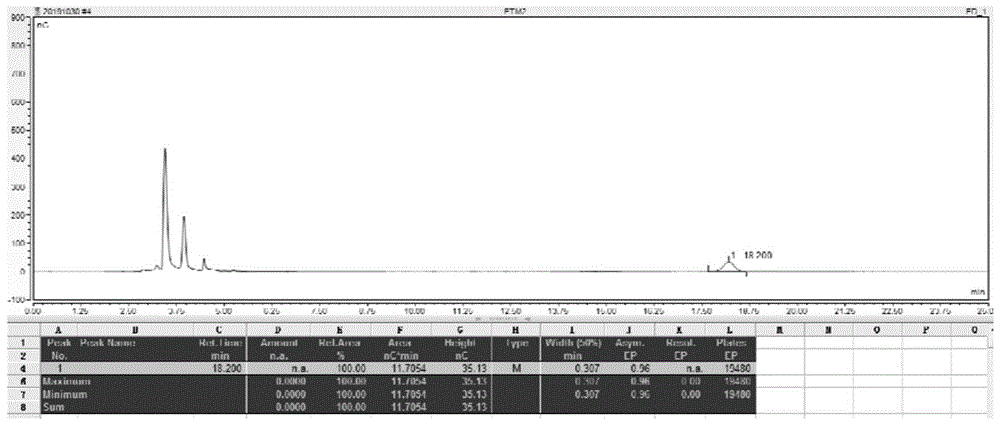
图1,1-n-乙基加洛糖胺ped检测图谱
图2,硫酸依替米星样品检测图谱
具体实施方案
通过以下具体实施例对本发明作进一步的说明,但不作为本发明的限制实施例1、1,3-n-乙基加洛糖胺
a、将依替米星原料药中加入30%双氧水进行破坏48h;
b、将破坏体系进行硅胶柱分离,得到1-n-乙基加洛糖胺;
c、在装有带干燥管的回流冷凝管的100ml干燥圆底烧瓶中,投入三乙胺10ml,乙酸酐2.5ml搅拌均匀后,加入1-n-乙基加洛糖胺1.7g,通过薄层色谱判断反应完全,改为蒸馏装置常压加热蒸出溶剂三乙胺。
d、降温至0~10℃,投入二氯甲烷10ml,然后在0~10℃条件下,滴加乙醛0.2ml,搅拌1h。加硼氢化钾0.5g,反应0.5h,加硼酸缓冲液3ml(1.0g硼酸加去离子水3.0ml搅拌溶解,用氢氧化钠调节ph=10),搅拌1.5h。
e、加入10%氢氧化钠搅拌0.5h,,常压加热蒸馏至液温100℃,蒸出部分溶剂。加入20%氢氧化钠溶液2ml,加热回流24h,降温至室温,真空抽虑得滤液。
f、滤液用无盐水稀释,通入层析分离柱中上样,用纯化水洗柱,40%乙醇洗脱有机相,浓缩。浓缩液通过制备液相根据药大方法进行分离,收集有效成分,浓缩得到1,3-n-乙基加洛糖胺,其纯度为98%,收率为80%。
取除盐浓缩液,用超纯水稀释浓度为10mg/ml,采用制备液相进行分离,收集有效成分。
所述制备液相,色谱条件如下:
实施例2、1,3-n-乙基加洛糖胺
a、将依替米星原料药中加入30%双氧水进行破坏48h;
b、将破坏体系进行硅胶柱分离,得到1-n-乙基加洛糖胺;
c、在装有带干燥管的回流冷凝管的100ml干燥圆底烧瓶中,投入乙腈10ml,乙酸酐2.5ml,搅拌均匀后,加入1-n-乙基加洛糖胺1.7g,通过薄层色谱判断反应完全,改为蒸馏装置常压加热蒸出溶剂三乙胺。
d、降温至0~10℃,投入二氯甲烷9ml,然后在0~10℃条件下,滴加乙醛0.2ml,搅拌1h。加硼氢化钾0.5g,反应0.5h,加硼酸缓冲液3ml(1.0g硼酸加去离子水3.0ml搅拌溶解,用氢氧化钠调节ph=10),搅拌1.5h。
e、加入10%氢氧化钠搅拌0.5h,,常压加热蒸馏至液温100℃,蒸出部分溶剂。加入20%氢氧化钠溶液2ml,加热回流24h,降温至室温,真空抽虑得滤液。
f、滤液用无盐水稀释,通入层析分离柱中上样,用纯化水洗柱,40%乙醇洗脱有机相,浓缩。浓缩液通过制备液相根据药大方法进行分离,收集有效成分,浓缩得到1,3-n-乙基加洛糖胺,其纯度为98%,收率为40%。
实施例3、1,3-n-乙基加洛糖胺
a、将依替米星原料药中加入30%双氧水进行破坏48h;
b、将破坏体系进行硅胶柱分离,得到1-n-乙基加洛糖胺;
c、在装有带干燥管的回流冷凝管的100ml干燥圆底烧瓶中,投入甲醇10ml,乙酸酐2.5ml搅拌均匀后,加入1-n-乙基加洛糖胺1.7g,通过薄层色谱判断反应完全。改为蒸馏装置常压加热蒸出溶剂三乙胺。
d、降温至0~10℃,投入二氯甲烷10ml,然后在0~10℃条件下,滴加乙醛0.2ml,搅拌1h。加硼氢化钾0.5g,反应0.5h,加硼酸缓冲液3ml(1.0g硼酸加去离子水3.0ml搅拌溶解,用氢氧化钠调节ph=10),搅拌1.5h。
e、加入10%氢氧化钠搅拌0.5h,,常压加热蒸馏至液温100℃,蒸出部分溶剂。加入20%氢氧化钠溶液2ml,加热回流24h,降温至室温,真空抽虑得滤液。
f、滤液用无盐水稀释,通入层析分离柱中上样,用纯化水洗柱,40%乙醇洗脱有机相,浓缩。浓缩液通过制备液相根据药大方法进行分离,收集有效成分,浓缩得到1,3-n-乙基加洛糖胺,其纯度为95%,收率为50%。
实施例4、1,3-n-乙基加洛糖胺的应用实施例,
1-n-乙基加洛糖胺的成功制备与分离,为计算硫酸依替米星原料药中1-n-乙基加洛糖胺的残留量提供依据。
举例如下:称取1,3-n-乙基加洛糖胺6.43mg,稀释至25ml,取定量的硫酸依替米星原料药稀释至25ml。根据以上数据及峰面积可以计算出硫酸依替米星原料药中杂质1,3-n-乙基加洛糖胺的残留量。
硫酸依替米星原料药的检测方法:
将供试品溶液和对照品溶液注入高效液相色谱仪,得到色谱图,根据色谱图计算依替米星原料药中杂质1,3-n-乙基加洛糖胺的残留量。
举例如下:称取1,3-n-乙基加洛糖胺6.56mg,稀释至25ml,取定量的硫酸依替米星原料药稀释至25ml。根据以上数据及峰面积可以计算出硫酸依替米星原料药中杂质1,3-n-乙基加洛糖胺的残留量。
色谱条件如下:
用十八烷基硅烷键合硅胶为填充剂(4.6mm×250mm,5μm或效能相当的色谱柱),以0.2mol/l三氟乙酸(含0.05%五氟丙酸,1.5g/l无水硫酸钠,0.8%(v/v)的50%氢氧化钠溶液,用50%氢氧化钠调节ph值至3.5)-乙腈(96:4)为流动相,柱温为35℃,流速为每分钟1.0ml,用积分脉冲安培电化学检测器检测,检测电极为金电极(推荐使用3mm直径),参比电极为ag/agcl复合电极,钛合金对电极,四波形检测电位,柱后加碱(50%氢氧化钠溶液1→25,推荐流速每分钟0.5ml)。
检测图谱见附图。
- 还没有人留言评论。精彩留言会获得点赞!