以TSPO为靶点的放射性分子探针及其制备方法和应用与流程
本发明涉及疾病诊断和治疗的放射性探针领域,尤其涉及一种含吡唑并嘧啶环系结构,靶向转位蛋白tspo阳性疾病的dpa714放射性分子探针的自动化制备方法,以及所述化合物作为疾病诊断的靶向放射性分子探针的临床应用。
背景技术:
转位蛋白tspo是一种由细胞核编码,内含169个氨基酸的疏水性蛋白(富含色氨酸),在各种族间高度保守,在组织中主要位于线粒体外膜,特别是在与甾体激素合成相关组织中高度表达,如唾液腺、性腺、肾上腺组织等,在心脏及肾脏组织中也有部分表达,而在脑和肝脏中表达水平很低。tspo是甾体合成的重要组成部分,可促进胆固醇跨膜转运进入磷脂膜,增加孕烯醇酮形成及下游神经类固醇合成,修复损伤神经和促进神经生长等。此外,tspo还参与许多生理功能,如线粒体呼吸凋亡、免疫应答和细胞增殖等。
在中枢神经系统中,tspo主要位于胶质细胞线粒体外膜,在静息小胶质细胞中水平表达较低。然而,当炎症反应发生时,大脑中小胶质细胞开始活化增殖,tspo密度显著增加,并且tspo增高密度与中枢神经系统炎症反应呈正相关:大脑损伤时,小胶质细胞和星形胶质细胞中tspo的表达上调,并与大脑损伤的程度直接相关;小胶质细胞的激活增殖并产生致炎性细胞因子如il-1、il-6、il-8、tnf-α和诱导型一氧化氮合酶(inos)等,引起脑内炎症反应,随着近年对多种神经系统病变如阿尔茨海默症、多发性硬化、脑梗、进行性肌肉萎缩症、帕金森病和亨廷顿舞蹈病等发病机制的深入探索,越来越多的研究结果表明,小胶质细胞激活介导的脑内慢性炎症反应(即tspo密度增高)是神经系统病变的病理特征之一。
作为一个潜在的药物靶点,tspo得到了越来越多研究者的关注。一方面,靶向tspo的pet显像技术逐渐成为评价脑损伤、诊断脑外伤进展与状态、区分外周炎症与肿瘤的重要工具;同时,该类正电子药物为神经退行性疾病等中枢神经系统疾病的早期诊断提供影像学研究依据。另一方面,各类特异性靶向tspo的配体在体内实验中已被证实对神经退行性病变和焦虑的动物模型有显著疗效,并具有神经保护、减少脑内炎症、促进神经再生、改善记忆和治疗应激相关障碍等功能。然而,在其治疗型药物的研究与开发中,仍有一些问题是值得探讨的,如在治疗局部神经适应证中tspo的中长期疗效;考虑到tspo在外周神经系统中高表达,长期给予tspo配体药物可能造成的不良反应等。而开发靶向型正电子药物可以对研究tspo配体的疗效和安全性提供有力支持。
第一个显像tspo的正电子药物是[11c](r)-pk11195,但由于其透脑率差、信噪比低等问题,无法满足临床的需求,越来越多的新型正电子药物被研发出来,具有代表性的包括[11c]pbr28、[11c]pbr111、[11c]daa1106、[11c]mbmp、[11c]er176、[18f]ab5186、[18f]fedaa1106、[18f]ge-180和[18f]dpa-714等,其中[18f]dpa-714以其高效便捷的放射标记方法、较低的非特异性结合以及较高的信噪比等优势,在多种疾病模型上进行了广泛的pet影像研究,包括脑创伤老鼠模型、慢性肝性脑病老鼠模型、亨廷顿病狒狒模型等,期望今后能在临床上应用于肌萎缩、中风后遗症以及阿尔兹海默症等神经炎症疾病的早期诊断和治疗疗效评估。
论文胡伟,赵军等,[18f]dpa-714的制备及其在动物体内的生物学分布《中华核医学与分子影像杂志》2017年第10期632-636页公开了一种[18f]dpa-714的制备方法,dpa-714以碳酸钾/k2.2.2溶液为相转移催化剂进行亲核取代反应.粗产物过铝柱预纯化,再经半制备hplc分离,检测产品在pbs和血浆中的稳定性,测定其脂水分配系数(logp),分别在小鼠和大鼠体内进行生物分布及micropet显像研究.结果[18f]dpa-714的放化产率为31.6%(未衰变校正),合成时间约为25min,放化纯≥99%;在pbs和血浆中放置4h,放化纯仍然≥99%.该标记化合物的logp为2.71,其药代动力学更符合二室模型,分布半衰期t1/2α为2.40min,消除半衰期t1/2β为69.15min;主要分布在肺和肾脏,心脏次之,注射后30min的放射性摄取值分别为(17.85±7.52)%id/g、(15.41±1.80)%id/g和(10.56±0.94)%id/g.该标记化合物主要通过肝脏代谢、肾脏排泄,骨的摄取较稳定.正常老龄大鼠60min动态扫描可见显像剂在脑实质内有摄取,清除缓慢.结论本实验合成的[18f]dpa-714具有标记率高、合成时间短、前体用量少等优点,并具有良好的生物分布和血-脑屏障通透特性。但是,该方法存在如下缺陷:(1)依靠实验人员手动操作,操作手法因人而异,可控性和重现性差,无法实现规范化操作;(2)因此制备的示踪剂质量(放射和化学纯度、溶剂残留、细菌和热原含量等)和批次之间的均一性无法保证,严格意义上讲,甚至不能满足动物实验要求;(3)这里用到的是放射性核素,放射性对人体是有严重损伤的,应该不接触或尽量少接触人体器官的,所有放射性核素操作的都是在铅屏蔽热室进行的,此处用到的手动制备中,手部和手臂等部分需完全暴露在放射性射线中,接受到的放射性较大,容易产生放射性损伤;(4)人体年最大接受放射性剂量是有上限的,因此制备次数和最大制备剂量很有限,因此,完成[18f]dpa-714的安全可靠、重现性好的自动化制备方法,是亟待解决的问题。
技术实现要素:
本发明的目的在于提供一种具有特异性靶向tspo的小分子有机化合物。
本发明的另一目的在于提供一种具有特异性靶向tspo的小分子有机化合物的制备方法。
本发明的另一目的在于提供一种以tspo为靶点的放射性分子探针。
本发明的另一目的在于提供一种以tspo为靶点的放射性分子探针的制备方法。
本发明的另一目的在于提供以tspo为靶点的放射性分子探针在制备tspo阳性病变显像诊断的放射性药物中的应用;所述tspo阳性病变为ad或脑卒中。
本发明是通过如下技术方案实现的:
本发明提供一种具有特异性靶向tspo的小分子有机化合物,如通式(i)所示:
式(i),
其中r为氢、溴基、对甲苯磺酰基、甲磺酰基、对硝基苯磺酰基或羟基;r连接的聚乙二醇(peg)的结构数量可以为0-5个,记为n=0-5。所述的化合物在本发明中记作r-pegn-dpa714。该结构中包含苯环结构、吡唑并咪唑结构和二乙基乙酰胺结构,所述苯环结构、吡唑并咪唑结构和二乙基乙酰胺基团分别通过c-c共价键连接;在苯环另一侧链上连接着r基团。
优选的,上述的具有特异性靶向tspo的小分子有机化合物,其结构式为:
r为氢、溴基、对甲苯磺酰基、甲磺酰基、对硝基苯磺酰基或羟基。
所述的化合物在本发明中记作r-dpa714。
更优选的,上述的具有特异性靶向tspo的小分子有机化合物,其结构式为:
上述的具有特异性靶向tspo的小分子有机化合物的制备方法,包括如下步骤:
a)将4-甲氧基苯甲酸甲酯与甲醇钠以摩尔比1:1的比例混合,在乙腈中加热回流24h,利用碱性催化消除反应得到3-(4-甲氧基-苯基)-3-氧基-丙腈为第一产物;
b)将步骤a)得到的第一产物在n,n-二乙基氯乙酰胺、碘化钠以摩尔比为1:1:3混合,加入氢氧化钠的乙醇溶液(80%),室温下反应7h,缩合得到3-氰基-n,n-二乙基-4-(4-甲氧基苯基)-4-氧基-丁酰胺为第二产物;所述乙醇溶液的质量百分比浓度为80%;
c)将步骤b)得到的第二产物与水合肼以摩尔比为1:2混合,加入少量醋酸溶解在乙醇中,加热至回流反应4h,得到2-[3-氨基-5-(4-甲氧基苯基)-1h-吡唑4-基]-n,n-二乙基乙酰胺为第三产物;
d)步骤c)得到的第三产物与2,4-戊二酮以摩尔比为1:1混合,在乙醇中加热至回流12h,得到n,n-二甲基2-[2-(4-甲氧基苯基)-5,7-二甲基吡唑[1,5-α]并嘧啶-3-基]乙酰胺为第四产物;
e)将骤d)得到的第四产物与三丁基十六烷基溴化磷以摩尔比为1:0.1混合,在45%氢溴酸溶液中加热至100℃7h,得到n,n-二甲基-2-[2-(4-羟基苯基)-5,7-二甲基-吡唑[1,5-α]并嘧啶-3-基]乙酰胺为第五产物,记为dpa713;
f)将骤e)得到的第五产物与氢化钠、对甲苯磺酰氯以摩尔比为1:1.1:2混合,加热至回流16h,得到n,n-二甲基-2-[2-[4-(2-对甲苯磺酰氧基乙氧基)苯基]-5,7-二甲基-吡唑[1,5-α]并嘧啶-3-基]乙酰胺为第六产物,记为tso-dpa714。
上述的具有特异性靶向tspo的小分子有机化合物的制备方法,简便易行,产物稳定性更高。
本发明的放射性标记前体tso-dpa714是以4-甲氧基苯甲酸甲酯为原料,经过了6步反应制得,反应简单、条件温和、收率稳定。
一种以tspo为靶点的放射性分子探针,它是放射性核素标记的有机小分子配合物,所述有机小分子是上述的具有特异性靶向tspo的小分子有机化合物。优选化合物:
所述的放射性核素选自[11c]或[18f],进一步优选[18f]。放射性核素18f为h218o经过回旋加速器轰击而成。
上述的以tspo为靶点的放射性分子探针,它是放射性核素18f标记的小分子配合物,所述有机小分子是具有特异性靶向tspo的小分子有机化合物,其结构式如下:
所述的放射性分子探针简化地表示为[18f]dpa714。
上述的以tspo为靶点的放射性分子探针的制备方法,包括以下步骤:
将tso-dpa714溶于无水乙腈中;用ge回旋加速器通过18o(p,n)18f核反应生产的放射性18f负离子,通过watersqma阴离子固相萃取柱浓缩;通过使用k222/k2co3的乙腈-水混合溶液淋洗至反应瓶中;鼓氮气加热直至溶液蒸干;加入溶有r-dpa714的乙腈溶液,加热至85℃保持10min;冷却后将反应溶液转入制备hplc中进行分离,分取产物峰通过watersc18固相萃取;c18柱再通过水洗除去水溶性杂质,用乙醇将吸附在c18柱上的产物通过无菌滤膜(0.22μm,13mm)过滤至无菌产品瓶中,并加入无菌注射用水稀释,使产物溶液中的乙醇浓度不高于10%,即得。
[18f]dpa714放射性检测方法,包括:
分析性hplc方法为使用高效液相色谱(美国waters公司,515型泵);紫外检测器(486型,uv吸收波长=254nm),放射性检测器(美国eg&gberthold公司),waters色谱柱nova-pakc18(4.6×150mm,5μm);
进一步的,上步中分析型hplc方法为使用watershplc系统配备watersc18分析柱(4.6mm×250mm),hplc梯度洗脱条件:乙腈/水(50/50,v/v),流速为1ml/min。从放射性hplc图谱中观察,[18f]dpa714的保留时间(retentiontime)为6.7min,放射化学纯度(radiochemicalpurity)>99%。
本发明通过使用getracerlabfx2n多功能合成仪,对tso-dpa714完成了自动化标记过程,通过一步亲核氟化反应和半制备hplc分离完成了放射性探针[18f]dpa714的自动化合成,合成过程简单、收率稳定、减少了操作人员所受到的放射性辐射。
本发明的以tspo为靶点的放射性分子探针的制备方法,通过苯环侧链上的对甲苯磺酰基作为标记位点,以无载体放射性氟-18作为放射源,设计并利用了温和的碱性反应条件,一步得到目标化合物[18f]dpa714,并通过半制备hplc分离程序分离得到高纯度的产品。
上述的以tspo为靶点的放射性分子探针在制备tspo阳性病变显像诊断的放射性药物中的应用;所述tspo阳性病变为ad或脑卒中。
放射性探针[18f]dpa714在体内可靶向tspo受体浓聚到病变部位,利用核医学的正电子断层显像技术,对tspo高表达病变进行显像诊断和治疗。
本发明有益效果为:
1、本发明提供一种具有特异性靶向tspo的小分子有机化合物,该有机化合物为有机小分子药物,以吡唑并嘧啶为母环,使其能够特异性靶向tspo受体,达到优异的神经炎症显像效果;在苯环与核素之间引入peg分子,以改善药代动力学特性,尤其是从非病灶组织的清除速率,以及调节亲脂性,控制药物在血脑屏障中的通透能力。
2、本发明的放射性标记前体tso-dpa714是以4-甲氧基苯甲酸甲酯为原料,经过了6步反应制得,反应简单、条件温和、收率稳定。
3、本发明的以tspo为靶点的放射性分子探针的制备方法,通过使用getracerlabfx2n多功能合成仪,对tso-dpa714完成了自动化标记过程,通过一步亲核氟化反应和半制备hplc分离完成了放射性探针[18f]dpa714的自动化合成,合成过程简单、收率稳定、减少了操作人员所受到的放射性辐射。
4、本发明的靶向转位蛋白tspo放射性示踪剂采用自动化制备,不用接触人体,保证了对人体的安全,另外,自动化制备,操作的最大剂量不受限制,重现性好,制备的示踪剂质量稳定,不仅可以满足动物试验的要求,还可直接用于临床。
5、本发明的以tspo为靶点的放射性分子探针的制备方法,(1)使用ge合成器完成自动化制备,之前无人报道;(2)自动化合成工艺成熟稳定,可控性、重现性较好,可实现规范性操作;(3)制备的示踪剂质量可控,每次制备完后自动生成生产记录,有可追溯,并能够保证批次之间的均一性;(4)整个合成过程通过合成器自动化完成,合成器放置在铅屏蔽热室内,操作员在外部使用电脑控制,整个过程无暴露机会,最大限度降低了人体的辐射吸附剂量。
附图说明
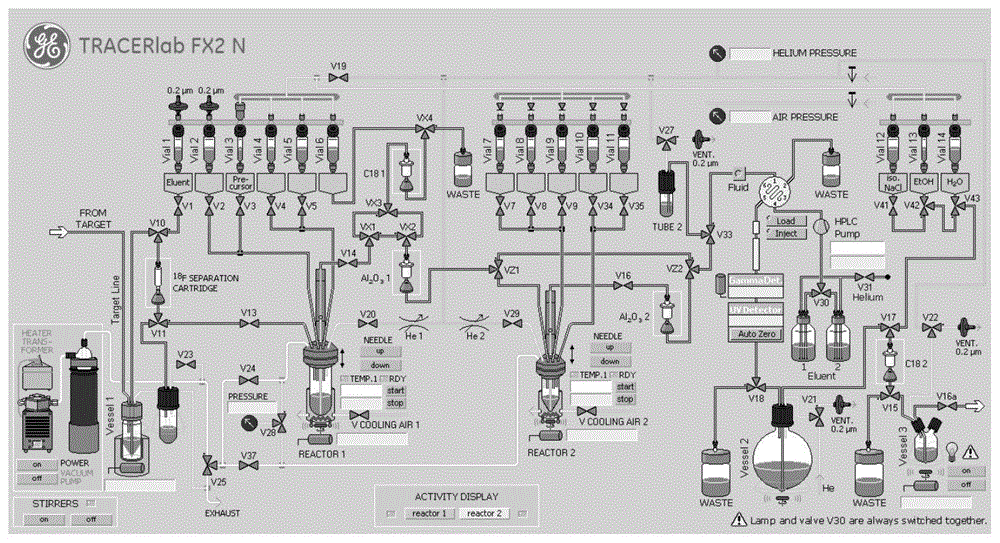
图1为getracerlabfx2n合成器的操作界面示意图。
图2为[18f]dpa714合成过程中半制备hplc的紫外(上)和放射(下)图谱。
图3为[18f]dpa714和标准品[19f]dpa714混合后进行hplc分析,得到的放射(上)和紫外(下)图谱。
图4为本发明的[18f]dpa714在脑出血患者中的显像结果,箭头指向病灶。
具体实施方式
以下结合附图及实例对本发明做进一步说明。
本发明实施例中所用仪器ge回旋加速器和getracerlabfx2n合成器均购自美国ge公司。
实施例1
一种具有特异性靶向tspo的小分子有机化合物,如通式(i)所示:
式(i),
其中r为氢、溴基、对甲苯磺酰基、甲磺酰基、对硝基苯磺酰基或羟基;r连接的聚乙二醇(peg)的结构数量可以为0-5个,记为n=0-5。所述的化合物在本发明中记作r-pegn-dpa714。其结构中包含苯环、吡唑并咪唑和二乙基乙酰胺结构,它由所述的三种母体结构通过共价键连接而成,其中核素标记位点位于苯环对位侧链上,并通过peg链与苯环连接,记为
优选的,上述的具有特异性靶向tspo的小分子有机化合物,其结构式为:
r为氢、溴基、对甲苯磺酰基、甲磺酰基、对硝基苯磺酰基或羟基。
所述的化合物在本发明中记作r-dpa714。
更优选的,上述的具有特异性靶向tspo的小分子有机化合物,其结构式为:
tso-dpa714的制备方法步骤如下:
1)化合物3-(4-甲氧基-苯基)-3-氧基-丙腈的合成:
搅拌下将30g4-甲氧基苯甲酸甲酯和9.75g甲醇钠于50ml玻璃瓶中混合均匀,在氩气保护下边搅拌边逐滴加入16.5ml乙腈和19ml氯苯,混合溶解后搅拌加热至回流24h,然后终止反应后经过固相柱色谱分离、浓缩后,得到第一产物5.16g,产率17%,产品为亮黄色晶体,熔点为132-137℃。1hnmr(cdcl3,300mhz)δ3.89(s,3h,och3),4.03(s,2h,ch2),6.97(d,j=9.0hz,2h,ph),7.90(d,j=9.0hz,2h,ph)。
2)化合物3-氰基-n,n-二乙基-4-(4-甲氧基苯基)-4-氧基-丁酰胺的合成:
将2.0g步骤1)得到的化合物3-(4-甲氧基-苯基)-3-氧基-丙腈和1.7gn,n-二乙基氯乙酰胺以及5.1g碘化钠混合均匀,将以上混合溶液搅拌下加入至溶有0.5gnaoh的80ml80%乙醇水溶液中,混合物在室温下搅拌7h并以tlc监测反应进程。待反应结束后终止反应并通过固相柱色谱纯化、浓缩后,得到第二产物2.1g,产率64%。1hnmr(cdcl3,300mhz)δ1.06-1.30(m,6h,n(ch2ch3)2),2.85(dd,j=4.5,16.2hz,1h,ch2),3.21-3.43(m,5h:4h,n(ch2ch3)2:1h,ch2),3.90(s,3h,och3),4.89–5.02(m,1h,ch),6.98(d,j=8.7hz,2h,ph),8.05(d,j=9.0hz,2h,ph)。
3)化合物2-[3-氨基-5-(4-甲氧基苯基)-1h-吡唑4-基]-n,n-二乙基乙酰胺的合成:
在含有0.73g步骤2)所得2-[3-氨基-5-(4-甲氧基苯基)-1h-吡唑4-基]-n,n-二乙基乙酰胺溶于37ml乙醇,置于100ml玻璃反应瓶中,加入0.73g水合肼和0.73ml冰醋酸混合均匀,混合物搅拌加热至回流4h,终止反应后通过固相硅胶柱色谱纯化、浓缩后,经得到第三产物1.52g,产率68%,产品为黄色晶体,熔点为154.5-157.5℃。经核磁共振鉴定产物:1hnmr(cdcl3,300mhz)δ0.90-1.10(m,6h,n(ch2ch3)2),3.04-3.33(m,4h,n(ch2ch3)2),3.50(s,2h,ch2),3.85(s,3h,och3),6.98(d,j=8.7hz,2h,ph),7.32(d,j=9.0hz,2h,ph)。
4)化合物n,n-二甲基2-[2-(4-甲氧基苯基)-5,7-二甲基吡唑[1,5-α]并嘧啶-3-基]乙酰胺的合成:
将1.2g步骤3)所得2-[3-氨基-5-(4-甲氧基苯基)-1h-吡唑4-基]-n,n-二乙基乙酰胺溶于20ml乙醇溶液中,置于100ml玻璃反应瓶中,向溶液中加入0.4g2,4-戊二酮,将混合溶液加热至回流12h,终止反应后通过固相硅胶柱色谱纯化、浓缩后,得到第四产物1.37g,产率93%,产品为浅黄色晶体,熔点为120.5-123.5℃。经核磁共振鉴定产物:1hnmr(cdcl3,300mhz)δ1.09-1.22(m,6h,n(ch2ch3)2),2.54(s,3h,5-ch3),2.74(s,3h,7-ch3),3.39–3.51(m,4h,n(ch2ch3)2),3.85(s,3h,och3),3.91(s,2h,ch2),6.51(s,1h,h-6),6.98(d,j=9.0hz,2h,ph),7.76(d,j=9.0hz,2h,ph)。
5)化合物n,n-二甲基-2-[2-(4-羟基苯基)-5,7-二甲基-吡唑[1,5-α]并嘧啶-3-基]乙酰胺的合成:
将0.43g步骤4)所得n,n-二甲基2-[2-(4-甲氧基苯基)-5,7-二甲基吡唑[1,5-α]并嘧啶-3-基]乙酰胺与0.06g三丁基十六烷基溴化磷、6ml48%氢溴酸混匀后,搅拌加热至回流7h,终止反应后通过固相硅胶柱色谱纯化、浓缩后,得到第五产物0.22g,记为dpa713,产率54%,产品为象牙色晶体,熔点为242.5-247℃。经核磁共振鉴定产物:1hnmr(cdcl3,300mhz)δ1.06-1.18(m,6h,n(ch2ch3)2),2.54(s,3h,5-ch3),2.73(s,3h,7-ch3),3.34-3.51(m,4h,n(ch2ch3)2),3.96(s,2h,ch2),6.49(s,1h,h-6),6.79-6.82(d,j=8.7hz,2h,ph),7.61–7.64(d,j=8.4hz,2h,ph)。
6)化合物n,n-二甲基-2-[2-[4-(2-对甲苯磺酰氧基乙氧基)苯基]-5,7-二甲基-吡唑[1,5-α]并嘧啶-3-基]乙酰胺的合成:
将0.5g步骤5)所得n,n-二甲基-2-[2-(4-羟基苯基)-5,7-二甲基-吡唑[1,5-α]并嘧啶-3-基]乙酰胺与38mg氢化钠和9ml干燥四氢呋喃混匀,室温搅拌下逐滴加入溶有1,2-二对甲苯磺酰基乙二醇1.04g的二氯甲烷溶液2ml,搅拌下加热回流16h,终止反应后通过固相硅胶柱色谱纯化、浓缩后,得到第六产物0.6g,记为tso-dpa714,产率77%,产品为白色粉末。经核磁共振鉴定产物:1hnmr(cd2cl2,400mhz)δ7.82(d,2h,j=8.3hz,ph),7.72(d,2h,j=8.8hz,ph),7.39(d,2h,j=8.3hz,ph),6.88(d,2h,j=8.8hz,ph),6.55(s,1h,ph),4.37(dd,2h,j=6.2&4.4hz,och2),4.19(dd,2h,j=6.2&4.4hz,ch2ots),3.87(s,2h,ch2(c=o)),3.50(q,2h,j=7.1hz,n(ch2ch3)),3.38(q,2h,j=7.1hz,n(ch2ch3)),2.72(s,3h,7-ch3),2.53(s,3h,5-ch3),2.45(s,3h,phch3),1.22(t,3h,j=7.1hz,n(ch2ch3)),1.11(t,3h,j=7.1hz,n(ch2ch3))。
实施例2
一种以tspo为靶点的放射性分子探针,该分子探针含有苯环、吡唑并咪唑和二乙基乙酰胺结构和放射性核素18f,放射性核素18f的反应位点在苯环的侧链上,所述放射性分子探针为[18f]dpa714。
所述[18f]dpa714放射性分子探针的制备方法步骤如下:
(1)18f的生产和活化
使用ge回旋加速器通过18o(p,n)18f核反应生产的放射性18f负离子,将含有18f负离子的h218o通过watersqma阴离子固相萃取柱,将18f负离子浓缩在qma柱上;随后使用k222/k2co3(13mg/4.5mg)的乙腈/水(0.3ml/0.3ml)混合溶液将吸附在qma柱上的18f负离子淋洗至反应瓶中;加热、鼓氮气直至溶液被蒸干。此时残留在反应瓶底部的即为活化的k222/k18f混合物。
(2)[18f]dpa714的标记和分离纯化
向反应瓶中加入溶有tso-dpa714(5mg)的乙腈溶液(1.5ml),混匀后加热至85℃保持10min;冷却后将反应溶液转入半制备hplc中分离,根据放射性检测器图谱分取产物峰至含有注射用水的稀释瓶中,使用watersc18固相萃取对产物从稀释的水溶液中进行浓缩;将c18柱再用注射用水洗除去水溶性杂质,用乙醇将吸附在c18柱上的产物洗至中转瓶。
(3)[18f]dpa714注射液的配置
向中转瓶中加入无菌注射用水稀释注射液,将稀释后的溶液通过无菌滤膜(0.22μm,13mm)过滤转至无菌产品瓶中,使注射液中的乙醇含量不高于10%,即得[18f]dpa714注射液。
(4)质量控制
hplc分析条件:色谱柱为c18柱(4.6mm×250mm),流动相a为乙腈,流动相b为水,流速为1ml/min:流动相a/b50/50。紫外检测波长为254nm,柱温20℃。放射性检测应用hplc专用放射性探测器。hplc分析结果显示:[18f]dpa714的保留时间(retentiontime)为6.7min(图3),放化纯度>99%,无需进一步纯化。为了验证[18f]dpa714的身份,将标准品[19f]dpa714与[18f]dpa714混匀后共同注射入分析hplc,对比放射性和uv检测器的出峰时间,经过检测,放射性和uv检测器的出峰时间一致,确定为[18f]dpa714。
(5)体外稳定性测定
[18f]dpa714注射液中加入胎牛血清,分别在60和120min分别用hplc测定其放射化学纯度。经测定,放射化学纯度均高于99%。
实施例3
一种以tspo为靶点的放射性分子探针[18f]dpa714,通过使用getracerlabfx2n合成器(见图1),完成自动化制备,包括以下步骤:
18f的生产和富集
使用ge回旋加速器通过18o(p,n)18f核反应生产的放射性18f负离子,将含有18f负离子的h218o传输至左侧锥形中转瓶中,打开真空泵将18f负离子通过watersqma阴离子固相萃取柱,将18f负离子浓缩于qma柱上。
(1)18f的活化
将淋洗液(k222/k2co3/乙腈/水13mg/4.5mg/0.3ml/0.3ml)预装在vial1中,打开v1将吸附在qma柱上的18f负离子淋洗至反应瓶reactor1中;将reactor1加热至95℃、抽负压、鼓氮气直至反应瓶中的溶液被蒸干。此时残留在反应瓶底部的即为活化的k222/k18f混合物。
(2)[18f]dpa714的标记
将tso-dpa714(5mg)溶解在干燥的乙腈溶液(1.5ml)中,预装在vial3中,打开v3将前体的乙腈溶液加入至含有活化18f离子的reactor1中,混匀后加热至85℃保持10min,冷却至50℃,标记反应完毕。
(3)[18f]dpa714的分离(见图2)
冷却后将反应混合溶液转入tube2中,将乙腈/h2o(1.2/1.2ml)润洗reactor1并转入tube2中;将tube2中的混合溶液转入半制备hplc中进行分离,洗脱条件为60%乙腈水溶液,流速为16ml/min,uv=254nm;通过放射性检测器和紫外检测器反馈的图谱,分取产物峰至含有50ml注射用水的稀释瓶中(图2中的方框代表产物的放射峰);使用watersc18固相萃取柱将产品从水溶液中进行浓缩。
(4)[18f]dpa714的纯化
将c18柱再用预装在vial13中的10ml注射用水洗,除去水溶性杂质;用预装在vial12中的1ml乙醇将吸附在c18柱上的产物洗至产品瓶中;用预装在vial11中的水10ml润洗c18柱和管路至产品瓶中,将产品中的乙醇稀释至10%以下。
(5)[18f]dpa714注射液的配置
将合成器产品瓶中的溶液转出,经过无菌滤膜(0.22μm,13mm)过滤转至无菌产品瓶中,使注射液中的乙醇含量不高于10%,即得[18f]dpa714注射液。
(6)[18f]dpa714身份确定
[18f]dpa714和标准品[19f]dpa714混合后共同注射入分析hplc的图谱(图3)。
(7)[18f]dpa714在脑出血患者中的显像
将上述[18f]dpa714放射性探针进一步制成无色透明针剂用于显像实验或显像诊断,具体实验及效果如下:临床确诊左侧基底节脑出血6个月后的患者,[18f]dpa714pet/ct图像见图4,静脉注射本发明的[18f]dpa7145mci,30min后使用gediscovery710pet/ct采集头部位图像,30min/床位,采集1个床位。病灶显示清晰,背景极地、信噪比高;出血区域形成软化灶,周围及同侧丘脑示踪剂凝聚(图4)(病灶用箭头标示)。
- 还没有人留言评论。精彩留言会获得点赞!