通过外消旋体拆分使用非对映体酒石酸酯制备(4S)-4-(4-氰基-2-甲氧基苯基)-5-乙氧基-2,8-二甲基-1,4-二氢-1,6-萘啶-3-甲酰胺的方法与流程
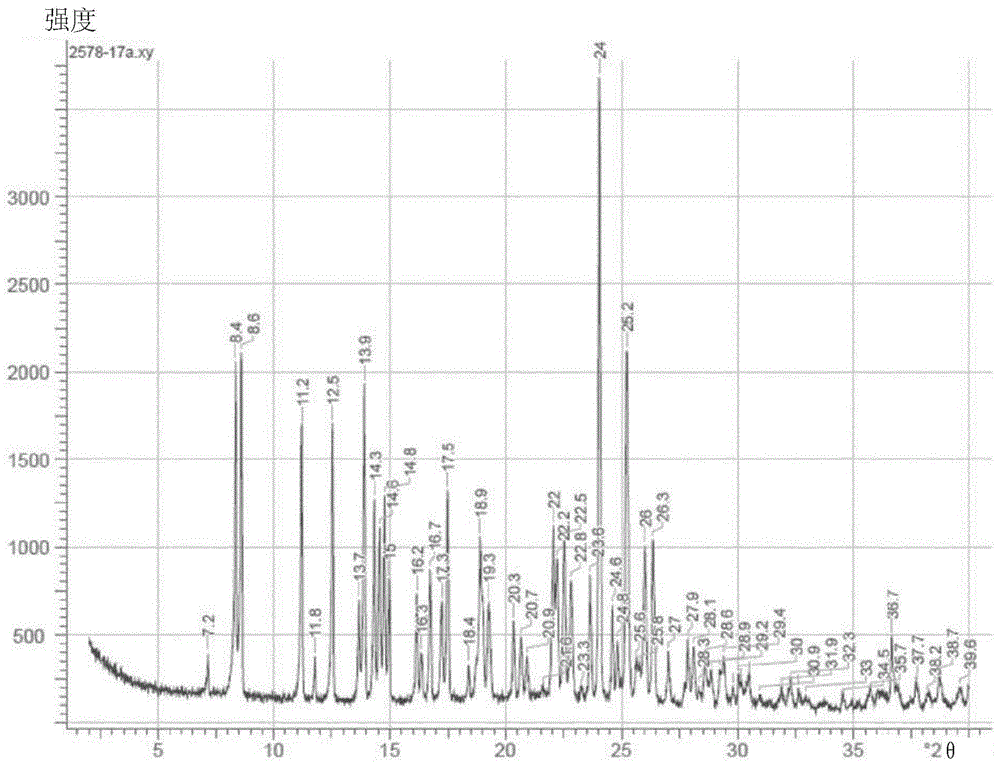
本发明涉及一种新的改进的制备式(i)的(4s)-4-(4-氰基-2-甲氧基苯基)-5-乙氧基-2,8-二甲基-1,4-二氢-1,6-萘啶-3-甲酰胺的方法以及涉及使用通式(iiia)和(iiib)的手性取代的酒石酸酯通过外消旋体拆分来制备对映体(ia)其中ar代表取代或未取代的芳族或杂芳族基团。芬尼酮(finerenone)(i)充当盐皮质激素受体的非甾体拮抗剂,并且可用作预防和/或治疗心血管和肾脏疾病例如心力衰竭和糖尿病肾病的药剂。式(i)的化合物及其制备方法记载于wo2008/104306和chemmedchem2012,7,1385以及wo2016/016287a1中。为了获得式(i)的化合物,必须将酰胺(ii)的外消旋混合物分离成对映体,因为仅式(i)的对映体具有活性。在公开的研究级合成(wo2008/104306)中,为此目的使用了专门合成的手性相(内部制备),其包含聚(n-甲基丙烯酰基-d-亮氨酸-二环丙基甲基酰胺)作为手性选择剂。已发现,还可以在容易商购获得的相上进行分离。其采用相chiralpakas-v,20μm的形式。所用洗脱液为甲醇/乙腈60∶40的混合物。在这种情况下,色谱分析可在常规色谱柱上进行,但优选使用本领域技术人员已知的技术,例如smb(模拟移动床;g.paredes,m.mazotti,journalofchromatographya,1142(2007):56-68)或varicol(computersandchemicalengineering27(2003)1883-1901)。尽管smb分离能够提供相对良好的收率和光学纯度,但是在gmp条件下这种设备的购置成本和操作的挑战巨大,同时伴随高的成本。所用的各个手性相也非常昂贵,并且仅具有有限的使用期限,在连续生产期间必须频繁更换。出于生产工艺的考虑,这不是最佳方法,除非存在第二家工厂以确保连续操作,这会带来额外的成本。此外,特别是在以吨级生产产品的情况下,溶剂回收是时间限制性步骤,需要购买巨大的降膜蒸发器,并消耗大量的能源。因此,本发明的一个目的是寻找用于分离对映体混合物的替代方案,该替代方案具有显著更高的成本效益并且可使用常规的中试装置(pilotplant)设备(搅拌式容器/分离装置)进行。所述设备为传统上制药生产厂的标准设备,并不需要额外的投资。此外,分批法的资格认定和验证比色谱方法的要容易得多,这是一个额外的优点。进行了许多尝试以开发使用外消旋体拆分的经典方法的分离(改变手性有机酸和溶剂),如表1中所示:表1:尤其是,我们还使用典型拆分剂(+)-酒石酸进行实验。然而,在任何情况下均未观察到盐形成;相反,在每种情况下,仅外消旋体从溶液中沉淀出,而未形成盐。这基本上符合本领域技术人员基于分子(ii)的pks而得到的预期,即,因为所测得的pks(对于碱而言)为4.3,这实际上排除了盐形成,因此通过与有机酸形成非对映体盐的经典外消旋体拆分应是不可能的,以至于盐形成只有在优选使用非常强的无机或有机矿物酸如手性磺酸或膦酸的情况下才是可能的。根据文献,例如“handbookofpharmaceuticalsalts-properties,selectionanduse;p.heinrichstahl,camilleg.wermuth(编辑);wiley-vch,第166页”,pk差应为至少3pk单位以使盐稳定形成。实际上,这是使用例如以下环状磷酸酯chlocyphos发现的:3当量的该环状磷酸酯与外消旋体(ii)的反应得到非对映体盐,其中(i)以仅44%e.e.的对映体过量存在。促使对映体过量趋向于>99%e.e.的所有努力均未成功;另外,chlocyphos不可大量商购获得;因此,我们研究了其他替代方案。在与烷基取代的酒石酸衍生物如(-)-o,o′-二新戊酰基-l-酒石酸或(-)-o,o′-二乙酰基-l-酒石酸的反应中未观察到盐形成。在对该主题的进一步研究中,出乎意料地,我们发现,酒石酸的芳族或杂芳族取代的衍生物非常适合于由外消旋体(ii)形成“非对映体盐”。因此,本申请提供(ii)的外消旋体拆分,其使用通式(iiia)或(iiib)的手性取代的酒石酸酯而拆分为(ia)和/或(i)其中ar代表未取代或取代的芳族或杂芳族基团。本发明还提供制备式(i)的(4s)-4-(4-氰基-2-甲氧基苯基)-5-乙氧基-2,8-二甲基-1,4-二氢-1,6-萘啶-3-甲酰胺的方法,其使用式(iiia)的手性取代的酒石酸酯通过(ii)的外消旋体拆分来进行其中ar代表未取代或取代的芳族或杂芳族基团。术语“取代”意指所述原子或基团上的一个或多个氢原子已被从指定的组中一个选择所替代,条件是在这种情况下不超过所述原子的正常价态。允许取代基和/或变量的组合。术语“未取代”意指没有氢原子被替代。杂芳基或杂芳族基团可为5元杂芳基,例如噻吩基、呋喃基、吡咯基、噁唑基、噻唑基、咪唑基、吡唑基、异噁唑基、异噻唑基、噁二唑基、三唑基、噻二唑基或四唑基;或6元杂芳基,例如吡啶基、哒嗪基、嘧啶基、吡嗪基或三嗪基;或三环杂芳基,例如咔唑基、吖啶基或吩嗪基;或9元杂芳基,例如苯并呋喃基、苯并噻吩基、苯并噁唑基、苯并异噁唑基、苯并咪唑基、苯并噻唑基、苯并三唑基、吲唑基、吲哚基、异吲哚基、吲哚嗪基(indolizinyl)或嘌呤基;或10元杂芳基,例如喹啉基(quinolinyl)、喹唑啉基、异喹啉基、噌啉基(cinnolinyl)、酞嗪基、喹喔啉基或蝶啶基。杂芳基特别为吡啶基、吡嗪基、吡咯基、吡唑基或嘧啶基。为了本申请的目的,芳基特别为苯基。用于本发明目的的合适的取代基为卤素、c1-c6-烷基、c1-c6-烷氧基、腈基、硝基、氰基、cf3基或酰胺基,所述酰胺基为例如nhcor,其中r代表甲基、乙基或苯基;或-nrcor基团,其中r具有上述含义;或conhr基团,其中r具有上述含义;或conrr′基团,其中r′具有与如上所定义的r相同的含义;或环酰胺如-co-吗啉基或-co-哌啶基。术语“卤素原子”是指氟、氯、溴或碘原子,优选氟、氯或溴原子。术语“c1-c6-烷基”表示具有1、2、3、4、5或6个碳原子的直链或支链的饱和一价烃基,例如甲基、乙基、丙基、异丙基、丁基、仲丁基、异丁基、叔丁基、戊基、异戊基、2-甲基丁基、1-甲基丁基、1-乙基丙基、1,2-二甲基丙基、新戊基、1,1-二甲基丙基、己基、1-甲基戊基、2-甲基戊基、3-甲基戊基、4-甲基戊基、1-乙基丁基、2-乙基丁基、1,1-二甲基丁基、2,2-二甲基丁基、3,3-二甲基丁基、2,3-二甲基丁基、1,2-二甲基丁基或1,3-二甲基丁基或其异构体。所述基团特别是具有1、2、3或4个碳原子(“c1-c4-烷基”),例如甲基、乙基、丙基、异丙基、丁基、仲丁基、异丁基或叔丁基,特别是1、2或3个碳原子(“c1-c3-烷基”),例如甲基、乙基、正丙基或异丙基。术语“c1-c6-烷氧基”表示式(c1-c6-烷基)-o-的直链或支链的饱和一价基团(其中术语“c1-c6-烷基”如上述定义),例如甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、仲丁氧基、异丁氧基、叔丁氧基、戊氧基、异戊氧基或正己氧基或其异构体。优选地,ar代表:其中r1、r2、r3、r4、r5各自代表氢原子或烷基,例如甲基、乙基、丙基;或卤素原子,例如氟、氯、溴或碘;或醚基,例如o-甲基、o-乙基、o-苯基;或硝基、氰基、cf3基或酰胺基,所述酰胺基为例如nhcor,其中r可代表甲基、乙基或苯基,或-nrcor基团,其中r具有上述含义,或conhr基团,其中r具有上述含义,或conrr′基团,其中r′具有与如上所定义的r相同的含义,或代表环酰胺如-co-吗啉基或-co-哌啶基。取代方式可能有很大的差异;因此,理论上可能有最高达5个不同的取代基,但通常优选单取代的ar基团。然而,ar也可为取代的杂芳族基团,例如优选吡啶或吡嗪。其也可为多环芳烃,例如取代的萘、蒽或喹啉。特别优选的ar为:其中*代表连接点。特别优选,ar代表:其中*代表连接点。非常特别优选的ar基团为:其中*代表连接点。其中,尤其优选未取代的环(苯基)。从文献中知晓酒石酸酯的制备,例如organicsynthesis,coll.第9卷,第722页(1998);第72卷,第86页(1995),以及chirality2011(23),3,第228页中所记载的。此外,本发明涉及下式的非对映体盐:其中ar代表未取代或取代的芳族或杂芳族基团,并具有上文给出的含义。特别优选其中ar代表苯基的非对映体盐。其实际上是典型的非对映体盐还是由氢键稳定的分子1∶1络合物,这是无法确定预测的。清楚的是,这些分子1∶1聚集体是非常稳定的,其行为类似于典型的非对映体盐,并且可被分离;因此,在下文中,我们仍使用术语非对映体盐。为了制备非对映体盐,使用通式(iiia)和(iiib)的酒石酸衍生物:其中ar代表取代或未取代的芳族或杂芳族基团。非对映体盐的制备如下进行:由外消旋混合物(ii)与通式(iiia)或(iiib)的酒石酸衍生物的反应得到的非对映体盐形成物具有4种可能性(iva-d)。出乎意料地,观察到某种优选,使得如果例如使外消旋体-(ii)与通式(iiia)的酒石酸衍生物反应,则在大多数情况下获得通式(iva)的非对映体盐,其中具有s构型的对映体优选形成盐。近乎定量地,非对映体盐(iva)从溶液中沉淀出,然后其可例如通过过滤从溶液中分离,而具有r构型的对映体保留在溶液中。以非常相似的方式,通过使外消旋体(ii)与通式(iiib)的酒石酸衍生物反应来制备通式(ivb)的镜像盐,其中具有r构型的对映体优选形成盐。沉淀的非对映体盐可近乎定量地分离出,并且在本文中所述s-对映体保留在溶液中。通过(ii)与(iiia)和(iiib)的化学计量比,并且通过选择溶剂,可以优化收率和对映体纯度。由于芬尼酮(i)具有s构型,因此s,s-构型的酒石酸衍生物优选用于外消旋体拆分,因为在这种情况下,优选形成s-对映体的非对映体盐。对于外消旋体拆分,使用0.4至1.2当量的酒石酸酯(iiia)或(iiib),优选0.4至0.7当量,特别优选0.45至0.6当量,非常特别优选0.50至0.55当量。非对映体盐的形成在由水和与水混溶的有机溶剂组成的溶剂混合物中进行。为了本申请的目的,合适的有机溶剂为例如乙醇、甲醇、异丙醇、1-丙醇、乙酸乙酯、异丁醇、二氯甲烷、1-戊醇或丙酮;然而,优选使用乙醇。所述溶剂还可以可商购获得的变性形式使用,例如通常用于乙醇的变性剂,例如甲苯、甲基乙基酮、噻吩、己烷,其具有很大的经济优势;因此,特别是对于工业规模的用途而言,可以使用酒精,其出于应用目的由任选地用甲苯或甲基乙基酮变性的乙醇组成。因此,为了本申请的目的,如果在下文中提及乙醇作为溶剂,则应理解为意指除纯的乙醇外,还具有上述定义意义上的酒精,特别是对于工业规模的用途而言。另外,还使用以下溶剂:乙酸乙酯/甲醇90∶10;甲醇/水80∶20;乙醇/水90∶10;乙醇/水85∶15;乙醇/水80∶20;乙醇/水75∶25;乙醇/水70∶30;二氯甲烷;1-丙醇/水80∶20;1-戊醇;1-戊醇/水90∶10;异丙醇;异丙醇/水80∶20;异丁醇/水90∶10;异丁醇/水80∶20;环己醇/水90∶10;苯甲醇/水90∶10;乙二醇;乙二醇/水80∶20。优选地,外消旋体拆分在乙醇/水混合物中进行,所述混合物(v/v)在乙醇∶水=1∶1至6∶1的范围内。然而,优选乙醇∶水=4∶1至3∶1的混合物。特别优选乙醇∶水=3∶1的混合物。该混合物可以预先制备或在将所有组分均已装入容器(pot)中后原位制备。基于外消旋体(ii)计,所述溶剂混合物可以过量10至40倍使用,例如每1kg外消旋体使用10l至40l溶剂混合物。优选过量10至20倍,特别是过量13至16倍,特别优选过量14至15倍。通常,外消旋体拆分通过下述方法进行:在室温下首先将所有组分加入到溶剂混合物中;然后加热至60℃至80℃,但优选至75℃;在75℃下搅拌2至10小时,优选3至4小时;然后在3至10小时、优选4至5小时(使用温度梯度(ramp))内冷却至室温(约20℃至23℃);然后将混合物在室温下再搅拌2至24小时,优选5至18小时,特别优选12至16小时。外消旋体拆分优选在75℃的温度下进行。随后,分离沉淀的非对映体盐(iva)、(ivb)、(ivc)和/或(ivd)。分离通过本领域技术人员已知的方法进行,例如通过过滤或使用离心机。以这种方式获得的滤饼可用溶剂或溶剂混合物洗涤一次或几次。然后在升高的温度(50℃至80℃,优选50℃)下在减压(优选<100毫巴)下干燥。在某些情况下,已发现使用载气是有利的。使用上述步骤,可以制备化学纯度非常高的非对映体盐。所述非对映体盐的对映体过量通常>97%e.e.(参见实施例)。非对映体盐不必一定要干燥,而是在下一个加工阶段中也可以使用湿的非对映体盐。除上述常规步骤之外,还可组合工艺步骤或改变其顺序,如下表2中所示:表2根据中试装置或生产中的设备类型,一种变型或其他变型可能是有利的。在下一步中,用碱处理非对映体盐并除去溶剂。溶剂的去除使用本领域技术人员已知的方法进行,例如通过蒸馏去除。为了制备手性化合物(i)和(ia),在蒸馏去除有机溶剂之后,必须用碱处理通式(iva)、(ivb)、(ivc)或(ivd)的非对映体盐,然后从溶液中沉淀出目标分子(i)或(ia)并将其分离——例如通过过滤和洗涤——然后式(iiia)或(iiib)的相应酒石酸酯以盐的形式保留在溶液中。为了本发明的目的,合适的碱为无机碱和有机碱。氨、氢氧化钠水溶液、氢氧化锂、氢氧化钾、碳酸铵、碳酸钠、碳酸钾、碳酸锂、碳酸氢铵、碳酸氢钠、碳酸氢钾、磷酸钠、磷酸钾、磷酸铵可用作无机碱。然而,优选使用氢氧化钠、磷酸钠或磷酸钾。特别优选使用磷酸钠或磷酸钾。必须强调的是,无机碱既可以无水形式使用也可以其水合物形式使用;因此,例如,可成功地使用磷酸钠(无水)和磷酸钠水合物。脂族或芳族碱例如三乙胺、咪唑、n-甲基咪唑、hünig碱、吡啶、1,8-二氮杂双环[5.4.0]十一碳-7-烯(dbu)可用作有机碱。目标化合物(i)或(ia)的释放在水和水混溶性有机溶剂的混合物中进行,所述水混溶性有机溶剂为例如乙醇、异丙醇、1,2-乙二醇、甲氧基乙醇、甲醇或丙酮;然而,优选使用乙醇。所述溶剂还可以市售的变性形式使用,例如用于乙醇的变性剂,例如甲苯、甲基乙基酮、噻吩、己烷,优选地,可使用酒精(spirits),其出于应用目的由任选地用甲苯或甲基乙基酮变性的乙醇组成,其具有很大的经济优势。已发现,使用水和乙醇的混合物是有利的,其中混合物(v/v)在乙醇∶水=1∶6至1∶3的范围内。然而,优选乙醇∶水=1∶4的混合物。该混合物可以预先制备或原位制备,在所有组分均已装入容器中时。这些混合物的用量可为所用非对映体盐(iva或ivb或ivc或ivd)的7至20倍,即,例如,1kg于7l至20l该混合物中。优选地,所述的混合物的用量为盐的量的8至12倍,特别优选9至11倍,非常特别优选10倍。目标化合物(i)或(ia)通过以下方式释放:在0℃至80℃、优选20℃至50℃下,首先将非对映体盐(iva或ivb或ivc或ivd)加入溶剂混合物中,然后加入有机或无机碱(以固体形式或作为溶液,优选于水中)以将ph调节至6.9至9.5、优选7.0至8.0、特别优选调节至ph7.5。任选地,所述碱可以非常快速地添加(在几分钟内),或非常缓慢地添加(在数小时内),例如在5分钟至3小时内。在任何情况下,优选更快速的添加;优选计量添加在5分钟至1小时内进行。为此,可使用安装在反应器中的ph计,其检查环境并缓慢添加碱。然而,还可以首先加入为确保达到所需的ph范围(根据经验)的固定量的碱(固体或溶解于溶剂中)。在制备中,这种步骤是特别优选的。已发现,在设定ph后,在10℃至80℃、优选20℃至60℃、优选40℃至60℃下继续搅拌是有利的。另外的搅拌时段可为1至10小时、优选2至5小时、特别优选3至4小时。然后将混合物冷却至15℃至25℃(例如使用斜坡),然后可再次搅拌1至64小时(该64小时用于证明所述方法的稳健性(robustness),参见实施例3b);然而,额外的搅拌时间优选为3至24小时,并且通常10至20小时就足够。分离通过本领域技术人员已知的方法进行,例如通过过滤或使用离心机。以这种方式获得的滤饼可用溶剂或溶剂混合物洗涤一次或几次。然后在升高的温度(50℃至80℃,优选50℃)下在减压(优选<100毫巴)下干燥。在某些情况下,已发现使用载气是有利的。然而,还可使用乙醇或水和乙醇的混合物溶解由过滤器获得的物质,然后在调节洗脱液中水/乙醇的量后,立即进行下一个工艺步骤(参见下文)。特别是当以工业规模作业时,该步骤是优选的,因为可省去干燥步骤。使用上述步骤,可以制备化学纯度非常高的粗产物。所述粗产物(i)和(ia)的对映体过量通常>96.5%e.e.。在扩大规模(up-scaling)过程中,发现从粗产物(i)和(ia)中完全去除酒石酸酯(iiia)和(iiib)在技术上并非总是简单,而且这些组分的含量接近规格界限(specificationlimit)。由于最终活性化合物的规格要求非常高(最终活性化合物中<0.1%(iiia)),因此已发现在某些情况下添加其他处理过程以确保酒石酸酯(iiia)的含量降低至低于0.15%、优选低于0.1%且特别是低于0.05%是有利的,并且最终活性化合物以符合规格的质量可再现地获得。在最终结晶化产生纯产物时,酒石酸酯(iiia)几乎没有消耗,或基本上完全没有消耗,因此,这样的后续处理过程确保整个过程的稳健操作,而且,该操作实际上无损耗。该处理过程,它确保了将活性化合物中的化合物(iiia)消耗至低于0.15%、优选低于0.1%且特别是低于0.05%,这也构成本发明主题的一部分。用于减少(iiia)的量的处理过程如下进行。用于所述处理过程的起始物料为(i)或(ia)的干燥的或有利地仍为湿的粗产物。再次,使用碱。无机或有机碱可用于除去式(iiia)或(iiib)的酒石酸酯。氨、氢氧化钠水溶液、氢氧化锂、氢氧化钾、碳酸铵、碳酸钠、碳酸钾、碳酸锂、碳酸氢铵、碳酸氢钠、碳酸氢钾、磷酸钠、磷酸钾、磷酸铵可用作无机碱。然而,优选使用氢氧化钠、磷酸钠或磷酸钾。特别优选使用磷酸钠或磷酸钾。必须强调的是,无机碱既可以无水形式使用也可以其水合物形式使用;因此,例如,可成功地使用磷酸钠(无水)和磷酸钠水合物。脂族或芳族碱例如三乙胺、咪唑、n-甲基咪唑、hünig碱、吡啶、dbu可用作有机碱。消耗(iiia)或(iiib)的操作是在水和水混溶性有机溶剂的混合物中进行,所述水混溶性有机溶剂为例如乙醇或异丙醇、1,2-乙二醇、甲氧基乙醇、甲醇、异丙醇或丙酮;然而,优选使用乙醇。所述溶剂还可以市售的变性形式使用,例如用于乙醇的变性剂,例如甲苯、甲基乙基酮、噻吩、己烷,其具有很大的经济优势。已发现,使用水和乙醇的混合物是有利的,其中混合物(v/v)在乙醇∶水=30∶10至10∶10的范围内。然而,优选乙醇∶水=20∶10的混合物。该混合物可预先制备或在所有组分均已装入容器中后原位制备。基于所用的非对映体盐(iva)、(ivb)、(ivc)或(ivd)计,可使用10至20倍量的溶剂混合物,即,例如,1kg于7l至20l该混合物中。优选地,所述混合物的用量为该混合物的量的10至14倍,特别优选11至12倍。消耗(iiia)或(iiib)的操作如此进行,即使得在40℃至80℃、优选50℃至70℃下首先将所述混合物加入到如上所述的溶剂混合物中,然后加入有机或无机碱(以固体形式或作为溶液,优选于水中)以将ph调节至6.9至9.5、优选7.5至9.0、特别优选调节至ph8.5。任选地,所述碱可以非常快速地(在几分钟内)添加,或非常缓慢地(在数小时内)添加,例如在1分钟至3小时内。在任何情况下,优选更快速的添加;优选计量添加在1分钟至1小时内进行。为此,可使用安装在反应器中的ph计,其可检查设定的ph并缓慢添加碱。然而,还可以首先加入为确保达到所需的ph范围(根据经验)的固定量的碱(固体或溶解于溶剂中)。在制备中,这种步骤是最优选的。已发现,在设定ph后,在40℃至80℃、优选50℃至75℃、优选60℃至70℃下继续搅拌是有利的。另外的搅拌时间可为1至24小时、优选2至10小时、特别优选2至4小时。然后将有机溶剂基本上蒸馏出;这可在大气压下进行或以更温和的方式在减压下进行。当有机溶剂已被基本上蒸馏出时,将混合物冷却至15℃至25℃(例如使用斜坡),然后可再次搅拌1至64小时(该64小时用于证明所述方法的稳健性,参见实施例3b);然而,额外的搅拌时间优选为1至24小时,并且通常1至5小时就足够。分离通过本领域技术人员已知的方法进行,例如通过过滤或使用离心机。以这种方式获得的滤饼可用溶剂或溶剂混合物洗涤一次或几次。然后在升高的温度(50℃至80℃,优选50℃)下在减压(优选<100毫巴)下干燥。在某些情况下,已发现使用载气是有利的。在蒸馏除去溶剂之前,还可使用以下物质将ph重新调节至ph5-7:有机酸,例如甲酸、柠檬酸、乙酸;或无机酸,例如盐酸、硫酸、磷酸;或酸性盐,例如磷酸二氢钠、硫酸氢钾或硫酸氢钠,然后如上所述进行。使用上述步骤,可以制备化学纯度非常高的粗产物。所述粗产物(i)和(ia)的对映体过量通常>97%e.e.。(iiia)或(iiib)的含量已降低至低于0.15%、优选低于0.1%且特别是低于0.05%。已发现,使以式(i)或(ia)的方式获得的粗产物另外结晶化以改善化学纯度且尤其是光学纯度是有利的,因为所述粗产物的对映体过量通常>97%e.e.。为此,研发出最终结晶化过程:由于gmp的原因,将粗产物(i)或(ia)溶于乙醇(如果需要则加热),然后进行颗粒过滤,优选使用酒精或用甲苯变性的乙醇。在某些情况下,取决于技术设备,所述结晶化也可在加压下、在升高的温度下进行(优点:溶剂较少);然而,也可从乙醇水溶液中重结晶(在高压或大气压下)。然后在大气压或减压下,将混合物浓缩至原始体积的约1/3至1/6、优选1/4至1/5;这引起产物的结晶。在某些情况下,例如,当必须通过较长时间将大量液体蒸馏出时,已发现在减压下进行蒸馏去除,保持内部温度较低以避免分解和副产物形成是有利的。蒸馏结束后,将混合物冷却至0℃,然后分离晶体,并在40℃至50℃下在减压下干燥。收率通常大于理论值的88%。根据ich指南,化学纯度和所达到的含量符合商业产品的标准。在中试批料中,残留溶剂含量——在当前情况下乙醇含量——通常<0.05%。光学纯度>>99%e.e.。在一个特别优选的方法中,特别是对于工业规模的操作,使用(+)-二苯甲酰基酒石酸(iii);可使用无水形式和水合物:外消旋体拆分优选在酒精/水混合物中进行。粗制芬尼酮的后续释放优选使用磷酸钠作为碱在酒精/水混合物中进行。在需要后处理的情况下,如果(+)-二苯甲酰基酒石酸(iii)的比例为>0.15%,则后处理优选使用磷酸钠作为碱在酒精/水混合物中进行。为得到纯的芬尼酮的最终结晶化优选使用酒精作为溶剂进行。本文中,获得0.15%、优选0.1%、特别优选小于0.05%杂质的纯度。还可从母液中分离目标对映体。本文中,首先,由(i)或(ia)制备合适的非对映体盐(iva)、(ivb)、(ivc)或(ivd),然后通过过滤进行分离,然后通过添加碱将包含相应的其他非对映体的母液的ph调节至ph>7、优选ph7.5,所述碱为例如氨、氢氧化钠水溶液、氢氧化锂、氢氧化钾、碳酸铵、碳酸钠、碳酸钾、碳酸锂、碳酸氢铵、碳酸氢钠、碳酸氢钾、磷酸钠、磷酸钾、磷酸铵,优选氢氧化钠、磷酸钠和磷酸钾,特别优选磷酸钠和磷酸钾。然后在大气压下或更温和地在减压下蒸馏出有机溶剂,优选乙醇;这引起相应的其他对映体沉淀。将产物滤出,用水或水/溶剂混合物洗涤并干燥。例如如实施例1c中所记载,从酒精中的适当地最终结晶化,得到适当纯的形式的化合物(i)和(ia)。因此,本申请还提供二苯甲酰基酒石酸含量≤0.15%的芬尼酮(i),其通过以下方式获得:使外消旋体(ii)与式(iii)的二苯甲酰基酒石酸在酒精/水混合物中反应以得到非对映体盐(vi)随后同样在酒精/水混合物中,使用磷酸钠来释放芬尼酮(i),然后在酒精/水混合物中再次与磷酸钠反应,然后使二苯甲酰基酒石酸含量≤0.15%的纯的芬尼酮结晶;在一个特别优选的实施方案中,获得二苯甲酰基酒石酸含量≤0.1%、特别是小于0.05%的芬尼酮。实验部分缩写和首字母缩写:etoh乙醇db酒石酸二苯甲酰基酒石酸dmso二甲基亚砜hplc高压高效液相色谱1h-nmr1h核磁共振波谱it内部温度ms质谱rt保留时间rrt相对保留时间tfa三氟乙酸tm覆盖温度(mantletemperature)xrpdx射线粉末衍射酒精用2%甲苯变性的乙醇实施例下表3示出了在hplc中回收的化合物的结构。hplc中保留时间的归属如下所示。表31)在二苯甲酰基酒石酸阶段用于检验杂质含量和对映体纯度的分析方法2)在非对映体盐阶段用于检验杂质含量和对映体纯度的分析方法3)在粗芬尼酮(i)的阶段用于检验杂质含量和对映体纯度的分析方法。以下实施例中所述的对映体测定的测量值均根据方法b测定。为了进行比较,将某些值,尤其是在中试装置中制备的批料的值,再次使用方法a进行测量,并得出可比较的结果。在以下实施例中给出的关于终产物纯芬尼酮(i)的纯度和含量的hplc分析数据仅指产物中存在的含量>0.05%的杂质。这基本上为杂质e。上文列出的表中所示出的所有其他杂质通常<0.05%。这类杂质的结构通过从浓缩母液中的分离来确定。实施例1使用无水(+)-o,o-二苯甲酰基-d-酒石酸(iii)的实验室批料实施例1a酒石酸盐(iv)的制备在室温(约23℃)下,首先将250g(660.616mmol)芬尼酮(ii)外消旋体装入3500ml由乙醇(用甲苯变性)/水=75∶25(v/v)组成的混合物中。使用固体用漏斗添加130.2g(363.339mmol)(+)-o,o-二苯甲酰基-d-酒石酸(iii),随后用250ml由乙醇(用甲苯变性)/水=75:25(v/v)组成的混合物冲洗。在0.75小时内将所得悬浮液加热至内部温度为75℃,然后在该温度下搅拌3.0小时。随后,使用冷却斜坡,将混合物在5.0小时内冷却至23℃,然后在该温度下搅拌过夜(约16小时)。将悬浮液通过玻璃料滤出,用250ml由乙醇(用甲苯变性)/水=75∶25(v/v)组成的混合物冲洗一次。湿产量:334.7g。然后在50℃下在减压(<100毫巴)下将湿产物干燥过夜(约16小时)。产量:250.2g(理论值的100.08%)无色晶体粉末。分析结果:ms(eipos):m/z=379[m+h]+1h-nmr(400mhz,dmso-d6):δ=1.05(t,3h),2.12(s,3h),2.18(s,3h),3.82(s,3h),3.99-4.07(m,2h),5.39(s,1h),5,89(s,2h),6.60-6.84(m(宽信号),2h),7.14(d,1h),7.28(dd,1h),7.37(d,1h),7.55(s,1h),7,61(t,4h),7.69(s,1h),7,75(t,2h),8,04(d,4h),12.50-15.40(非常宽的信号,2h)以及溶剂dmso的信号和增强的水信号:δ=2.5-2.6,以及在δ=3.40-3.50(q)和δ=1.05-1.10(t)处的较小峰,来自残留溶剂乙醇的叠加信号。根据实施例1a获得的无色晶体粉末(芬尼酮-(+)-o,o-二苯甲酰基-d-酒石酸络合物(vi))在以下条件下于25℃下通过xrpd检测:样品制备:使粉末在两层膜(例如聚乙酸酯)之间制成的薄层仪器:x射线粉末衍射仪x′pertpro发生器:40kv/40ma探测器:pixcel辐射:cuka辐射波长:技术:传输扫描范围:2°£2q£40°步长:0.013°2q测量时间:25s/步结果示于图1和表1中。表1:实施例1b制备粗产物(i)在室温下,将248g实施例1a中制备的化合物(iv)悬浮于2480ml由乙醇(用甲苯变性)/水=20∶80(v/v)组成的混合物中(测定ph为ph=4)。随后,在60分钟内逐滴加入819.6g磷酸钠水溶液(100g磷酸钠溶解于1000ml水中),并将ph调节至ph=7.2。将混合物在23℃下再搅拌50分钟(ph=7.1)。随后,在10分钟内逐滴加入98.3g磷酸钠水溶液(100g磷酸钠溶解于1000ml水中),并将ph调节至ph=7.5。在1小时内,将混合物加热至内部温度为50℃,并在该温度下搅拌3.0小时。将混合物在1小时内冷却至22℃,并在该温度下再搅拌1小时。将晶体通过玻璃料滤出,并用200ml和100ml由乙醇(用甲苯变性)/水=20∶80(v/v)组成的混合物分别洗涤一次,以及用200g水洗涤两次。湿产量:263.4g。然后在50℃下在减压(<100毫巴)下将湿产物干燥整个周末(>48小时)。产量:116.9g(理论值的93.52%)无色晶体粉末。分析结果:ms(eipos):m/z=379[m+h]+1h-nmr(400mhz,dmso-d6):δ=1.05(t,3h),2.12(s,3h),2.18(s,3h),3.82(s,3h),3.99-4.07(m,2h),5.37(s,1h),6.60-6.84(m(宽信号),2h),7.14(d,1h),7.28(dd,1h),7.37(d,1h),7.55(s,1h),7.69(s,1h)以及溶剂dmso的信号和明显增强的水信号:δ=2.5-2.6,以及在δ=3.38(未归属)处很小的峰。实施例1c制备纯产物(i)将116.0g实施例1b中制备的粗产物(i)悬浮于2330ml乙醇(用甲苯变性)中,然后加热至回流。在加热下,产物溶解。在该温度下继续搅拌1小时。将溶液通过加热的压力过滤器(t=75℃)滤出,然后用30ml乙醇(用甲苯变性)冲洗压力过滤器。然后蒸馏出溶剂直至最终体积达到约4倍(相对于所用物质:116gx4~484ml)(蒸馏出约1920ml)。然后将混合物冷却至内部温度为23℃(持续时间约1.5至2小时)。然后将混合物在内部温度为3℃下搅拌2小时。将产物滤出并用100ml乙醇(用甲苯变性)冲洗一次。湿产量:124g。在50℃下在减压(<100毫巴)下将湿产物干燥整个周末(>48小时)。产量:112.6g(理论值的97.07%)无色晶体粉末,细针状晶体。分析结果:ms(eipos):m/z=379[m+h]+1h-nmr(400mhz,dmso-d6):δ=1.05(t,3h),2.12(s,3h),2.18(s,3h),3.82(s,3h),3.99-4.07(m,2h),5.37(s,1h),6.60-6.84(m(宽信号),2h),7.14(d,1h),7.28(dd,1h),7.37(d,1h),7.55(s,1h),7.69(s,1h)和溶剂dmso以及水在δ=2.5-2.6处的弱信号和在δ=3.38(未归属)处很小的峰。变型:moda(根据通过x射线确定绝对构型的wo2016/016287a1中的定义)实施例2使用(+)-o,o-二苯甲酰基-d-酒石酸水合物(iii)的实验室批料实施例2a酒石酸盐的制备在室温下首先装入350.0g外消旋体(ii),然后加入3112g酒精。然后加入1200g水。使用固体用漏斗添加191.4g(+)-o,o-二苯甲酰基-d-酒石酸一水合物(iii),随后用113g水冲洗。将悬浮液在1小时内加热至内部温度为75℃,然后在75℃下搅拌3小时。随后,使用冷却斜坡,将混合物在5.0小时内冷却至23℃,然后在该温度下搅拌过夜(约18小时)。将悬浮液通过玻璃料滤出,并用由332.3g酒精和140.2g水组成的混合物洗涤两次。湿产量:487.6g。然后在50℃下在减压(<100毫巴)下,将湿产物干燥整个周末(>48小时)。产量:351.0g(理论值的100.29%)无色晶体粉末。以类似于实施例1b和1c中所述的方式,纯芬尼酮(i)可由该酒石酸盐制备。实施例3以2kg外消旋体(ii)开始,在中试装置中制备3批纯芬尼酮(i)。实施例3a酒石酸盐(iv)的制备在室温(约23℃)下,首先将2.00kg(5.285mol)外消旋体芬尼酮(ii)加入到28.0l由乙醇(用甲苯变性)/水=75∶25(v/v)组成的混合物中。使用固体用漏斗添加1.042kg(2.907mol)(+)-o,o-二苯甲酰基-d-酒石酸(iii),随后用2l由乙醇(用甲苯变性)/水=75∶25(v/v)组成的混合物冲洗。将所得悬浮液在45分钟内加热至内部温度为75℃,然后在该温度下搅拌3.0小时。随后,使用冷却斜坡,将混合物在5.0小时内冷却至23℃,然后在该温度下搅拌过夜(约16小时)。将悬浮液通过玻璃料滤出,用2l由乙醇(用甲苯变性)/水=75∶25(v/v)组成的混合物冲洗一次,并且使其被吸干10分钟。湿产量:2.69kg。然后在50℃下在减压(<100毫巴)下,将湿产物干燥直至质量保持恒定(约17小时后)。产量:2.009kg(理论值的~100%)无色晶体粉末。实施例3b制备粗产物(i)在室温(约23℃)下,将2.006kg实施例3a中制备的化合物(iv)悬浮于20.0l由乙醇(用甲苯变性)/水=1∶4(v/v)组成的混合物中(测定ph为ph=4)。然后逐滴加入6.05kg9.09%浓度的磷酸钠水溶液,并将ph调节至ph=7.2。将混合物在23℃下再搅拌约50分钟(ph=7.17)。然后逐滴加入0.65kg9.09%浓度的磷酸钠水溶液,并将ph调节至ph=7.5。在1小时内,将混合物加热至内部温度为50℃,并在该温度下搅拌3.0小时。将混合物在1小时内冷却至22℃,并在该温度下搅拌整个周末(约64小时*)。将晶体通过k800seitz过滤板滤出,并用1.6l和0.8l由乙醇(用甲苯变性)/水=20:80(v/v)组成的混合物分别洗涤一次,并且以每次1.6l水洗涤两次。湿产量:1.82kg。然后在50℃下在减压(<100毫巴)下将湿产物干燥直至质量保持恒定(约17小时后)。产量:0.978kg(理论值的97.8%)无色晶体粉末。*)由于技术原因,将混合物在整个周末搅拌;通常,搅拌14小时就足够。实施例3c制备纯产物(i)将250g实施例3b中制备的粗产物(i)悬浮于5000ml乙醇(用甲苯变性)中,然后加热至回流。在加热下,产物溶于溶液中。在该温度下继续搅拌1小时。将溶液通过加热的压力过滤器(t=85℃)滤出,然后用200ml乙醇(用甲苯变性)冲洗压力过滤器。然后蒸馏出溶剂直至最终体积达到约4倍(相对于所用物质:250gx4~1000ml)(蒸馏出约4200ml)。然后将混合物冷却至内部温度为4-5℃(斜坡:持续时间约4小时)。然后将混合物在内部温度为4-5℃下搅拌1小时。使产物滤出并用220ml乙醇(用甲苯变性)冲洗一次。湿产量:251.2g。在50℃下在减压(<100毫巴)下,将湿产物干燥整个周末(>48小时)。产量:223.0g(理论值的89.2%)无色晶体粉末。在每种情况下以2kg芬尼酮(ii)的外消旋体开始的3个批料的结果总结于下表4中:表4:酒石酸盐(iv)批料所用外消旋体(ii)kg产量:kg收率:理论值的%批料1(实施例3a)22.009~100批料222.025~100批料322.027~100粗产物(i)在所有3个批料中,所述(+)-o,o-二苯甲酰基-d-酒石酸含量为:0.00%。纯产物(i)批料粗产物(i):g产量:g收率:理论值的%批料1(实施例3c)25022389.2批料225023594.0批料325022691.8在所有3个批料中,所述(+)-o,o-二苯甲酰基-d-酒石酸含量(iii)为0.00%。实施例4方法的工业化实现。以下实施例描述了所述方法在工业规模上的实现。然后将2批的纯芬尼酮(i)的结果如产率和分析数据示于表中:实施例4a酒石酸盐(iv)的制备批料大小为80kg外消旋体芬尼酮(ii)。·在1600l搅拌式容器中,首先在it为20℃下装入711.0kg酒精,接着加入300kg水,然后加入80.0kg外消旋体(ii),并且最后加入41.7kg(+)-二苯甲酰基酒石酸(iii)。·将混合物加热至it为75℃,并搅拌3小时。·将混合物在5小时内(斜坡)冷却至it为23℃,并再搅拌16小时。·为了进行离心分离,将悬浮液转移至翻袋式离心机(invertingcentrifuge)中,进行离心分离,用由10.7kg酒精和4.5kg水组成的混合物冲洗,然后进行滚转干燥(tumble-dried)并移出。·在50℃的覆盖温度(mantletemperature)和在30毫巴下于300l球式干燥器中进行干燥。在45℃的内部温度下终止干燥(持续时间约6小时)。·将产物冷却至15℃,然后移出。实施例4b粗产物(i)批料大小为89.3kg酒石酸盐(iv)起始物料·制备磷酸钠溶液。·在630l搅拌式容器中,在20℃下首先加入360.0kg完全去矿物质水。·加入36.0kg磷酸钠并溶解。·在1600l搅拌式容器中,在20℃下首先加入141.1kg酒精。·然后加入714.2kg水和89.3kg酒石酸盐(iv)。·将混合物加热至内部温度为50℃。·然后计量加入295.1kg磷酸钠溶液·在1.5小时后,通过添加35.4kg磷酸钠溶液将ph调节至ph=7.5(+/-0.1)。·将混合物搅拌,并通过斜坡(3小时)冷却至it=22℃。·然后在it=22℃下继续搅拌(18小时)。·在接收器中,制备129.6kg水和25.6kg酒精的混合物(用于洗涤)。·将悬浮液计量加入翻袋式离心机中(约4次批料+剩余批料),并且每次少许地,用129.6kg水和25.6kg酒精的混合物洗涤一次,以及用288.0kg完全去矿物质水洗涤一次。·将产物移出并在球式干燥器中干燥(tm=50℃,30毫巴,在it=45℃下终止,约6小时)。·将产物冷却并在<30℃下移出。实施例4c粗产物(i)的再处理在某些情况下,如果粗产物中(+)-二苯甲酰基酒石酸(iii)的规格界限>0.1%,则已发现进行后处理(参见实施例6)是有利的,以确保该界限<0.1%可重复实现。另外,后处理过程实际上是无损耗的,并且通常以>95%的收率得到粗产物。再处理的粗产物(i)批料大小为30kg粗产物(i)。·磷酸钠溶液的制备:在室温下,在250l搅拌式接收器中将2.3kg磷酸钠溶解于147.8kg水中。·在20℃下,首先在630l搅拌式容器中加入198.8kg酒精,并加入120kg水,然后加入30kg粗产物(如表5中所述含有(+)-二苯甲酰基酒石酸(iii))。·将混合物加热至内部温度为70℃,并搅拌30分钟。·在70℃下,计量加入14.1kg上述制备的磷酸钠溶液,将ph调节至ph8.9(+/-0.1),并将混合物在70℃下搅拌20小时。·使用斜坡,将混合物在60分钟内冷却至44℃,并加入300g纯芬尼酮(i)晶种。·然后在200毫巴下且内部温度为最高60℃下蒸馏出溶剂,同时将369kg水计量加入到搅拌式容器中。·最后,将混合物在1.5小时内冷却至22℃,并将产物在加压式吸滤器上分离,并且分为三份,用总共180l水洗涤。·然后在50℃的覆盖温度下且在减压(30毫巴)下,将产物干燥至重量恒定。·将产物冷却至15℃,然后取出。实施例4d纯产物(1)批料大小为26kg(2次13kg)经再处理的粗产物(i)·在250l搅拌式容器中,在20℃下首先将13kg经再处理的粗产物(i)加入到184.9kg酒精中,所述搅拌式容器已被预先惰性化。·将溶液加热至80℃的覆盖温度,并在78℃下搅拌30分钟(伴随着产物溶解)。·使溶液通过0.65μm滤芯(pp元件sartorius)过滤(gmp过滤),并将滤液转移至630l搅拌式容器中。·产物用18.5kg酒精洗涤。·对于2个细分部分(2次13kg产物)重复上述操作,并将溶液合并。·在大气压下蒸馏出溶剂至最终装料高度(fillinglevel)为约130l(分两部分,覆盖温度:104℃,持续时间约6小时)。·使用线性温度斜坡将溶液在4小时内冷却至0℃,然后再搅拌1小时。·将产物悬浮液在加压式吸滤器上分离,并用45.9kg酒精洗涤。·然后在50℃下在减压(30毫巴)下将湿产物干燥至重量恒定(约12小时)。将混合物冷却至15℃并将产物移出。用于制备纯芬尼酮(i)的3个工业批料的收率和分析结果的总结在表5中给出。酒石酸盐(iv)表5:粗产物(i)再处理后的粗产物(i)批料输入(i):kg产量(i):kg收率:理论值的%批料1(实施例4c)3029.899.33批料23029.899.33纯产物,纯芬尼酮(i)批料输入kg产量:kg收率:理论值的%批料1(实施例4d)2623.188.85批料22622.988.08实施例5类似于实施例1b的过程的几种变型适用于释放粗芬尼酮(i)。本文中,目的是保持粗产物中的(+)-o,o-二苯甲酰基-d-酒石酸(iii)的比例尽可能小(0.15%或更小)。具体地,对于计量加入磷酸钠溶液后的最终ph、温度、计量加入磷酸钠溶液的时间和另外的搅拌时间进行改变,并且研究了它们对于粗产物中的(+)-o,o-二苯甲酰基-d-酒石酸的含量的影响。批料大小:100g酒石酸盐(iv)于158.0g酒精和799.8g水中所用酒石酸盐(iv)的分析数据:结果总结于下表6中:表6实施例6再处理方法实施例6a经过再处理的粗产物:(+)-o,o-二苯甲酰基-d-酒石酸的比例降低<0.1%该实验通过有目的地使用含有升高比例的(+)-o,o-二苯甲酰基-d-酒石酸(iii)的批料来证明再处理方法的稳健性。根据分析,将仍含有0.77%的(+)-o,o-二苯甲酰基-d-酒石酸(iii)的粗产物用于再处理。粗产物的再处理:(+)-o,o-二苯甲酰基-d-酒石酸(iii)的比例降低<0.1%根据分析,将仍含有0.77%的(+)-o,o-二苯甲酰基-d-酒石酸(iii)的粗产物用于再处理。采用以下过程:将150g粗产物(i)(含有0.77%的(+)-o,o-二苯甲酰基-d-酒石酸(iii))加入到600g水和953.9g酒精中,并加热至70℃。在一个小时内,全部固体溶解。然后将混合物在70℃下搅拌30分钟。ph为ph=5.0。通过加入44.6g磷酸钠水溶液(每985g水15g磷酸钠),将ph调节至ph=8.5,然后将混合物在70℃下搅拌2小时。在30分钟内,将混合物冷却至53℃,并给予纯物质(晶种)作为种子。然后在50-52℃下继续搅拌30分钟。在200至155毫巴和内部温度下,将乙醇几乎完全蒸馏出,同时计量加入1845ml水(装料高度保持恒定:蒸馏出的量=添加的水量)。关闭加热,并且在23℃下将悬浮液搅拌过夜。晶体经由玻璃料滤出,并且以每次300ml水洗涤三次。湿产量:218.9g。在50℃下在减压(<100毫巴)下将湿产物干燥整个周末(>48小时)。产量:146.7g(理论值的97.8%)。实施例6b证明稳定性,在70℃下搅拌20小时粗产物的再处理:(+)-o,o-二苯甲酰基-d-酒石酸(iii)的比例降低<0.1%根据分析,将仍含有0.77%的(+)-o,o-二苯甲酰基-d-酒石酸(iii)的粗产物用于再处理。采用以下过程:将150g粗产物(i)(含有0.77%的(+)-o,o-二苯甲酰基-d-酒石酸(iii))加入到600g水和953.9g酒精中,并加热至70℃。在一个小时内,全部固体溶解。然后将混合物在70℃下搅拌30分钟。ph为ph=5.0。通过加入45.2g磷酸钠水溶液(每985g水15g磷酸钠),将ph调节至ph=8.5,然后将混合物在70℃下搅拌20小时。在30分钟内,将混合物冷却至53℃,并给予纯物质(晶种)作为种子。然后在50-52℃下继续搅拌30分钟。在200至155毫巴和内部温度下,将乙醇几乎完全蒸馏出,同时计量加入1845ml水(装料高度保持恒定:蒸馏出的量=添加的水量)。关闭加热,并且在23℃下将悬浮液搅拌过夜。晶体经由玻璃料滤出,并且以每次300ml水洗涤三次。湿产量:194.3g。在50℃下在减压(<100毫巴)下将湿产物干燥过夜(约16小时)。产量:143.1g(理论值的95.4%)。实施例6c粗产物的再处理:(+)-o,o-二苯甲酰基-d-酒石酸(iii)的比例降低<0.1%。使用磷酸二氢钠将ph反滴定至ph7.0。根据分析,将仍含有0.77%的(+)-o,o-二苯甲酰基-d-酒石酸(iii)的粗产物用于再处理。采用以下过程:将375.0g粗产物(i)(含有0.77%的(+)-o,o-二苯甲酰基-d-酒石酸(iii))加入到1500g水和2384.8g酒精中,并加热至70℃。在一个小时内,全部固体溶解。然后将混合物在70℃下搅拌30分钟。ph为ph=4.9。通过加入101.4g磷酸钠水溶液(每985g水15g磷酸钠),将ph调节至ph=8.5,然后将混合物在70℃下搅拌20小时。随后,使用磷酸二氢钠溶液(50g磷酸二氢钠于200ml水中)将ph调节至ph=7。在30分钟内,将混合物冷却至53℃,并给予纯物质(晶种)作为种子。然后在50-52℃下继续搅拌30分钟。在200至155毫巴和内部温度下,将乙醇几乎完全蒸馏出,同时计量加入4610ml水(装料高度保持恒定:蒸馏出的量=添加的水量)。最后,在90分钟内将混合物冷却至23℃。然后在22℃下将反应混合物搅拌整个周末(约70小时)。晶体经由玻璃料滤出,并且以每次750ml水洗涤三次。湿产量:402.3g。在50℃下在减压(<100毫巴)下将湿产物干燥过夜(约16小时)。产量:336.6g(理论值的89.76%)。实施例6d来自实施例6c的批料的最终结晶化将80.0g实施例6c中制备的粗产物(i)悬浮于1600ml乙醇(用甲苯变性)中,然后加热至回流。在加热下,产物溶解。在该温度下继续搅拌1小时。将溶液通过加热的压力过滤器(t=75℃)滤出,然后用100ml乙醇(用甲苯变性)冲洗压力过滤器。然后在减压下(覆盖温度45-47℃)尽可能温和地蒸馏出溶剂,直至最终体积达到约5倍(相对于所用物质:80gx5~400ml)。然后,在1小时内,将混合物冷却至内部温度为2℃,并在该温度下搅拌1小时。将产物滤出并用80ml乙醇(用甲苯变性)冲洗一次。湿产量:83.3g。在50℃下在减压(<100毫巴)下将湿产物干燥过夜(约16小时)。产量:71.4g(理论值的89.3%)无色晶体粉末,细针状晶体。分析结果:实施例7芬尼酮对映体(ia)的分离实施例7a(-)-o,o-二苯甲酰基-l-酒石酸盐的制备在室温下首先加入187.2g外消旋体(ii),然后加入1662.5g酒精。然后加入485.5g水。使用固体用漏斗添加97.5g(-)-o,o-二苯甲酰基-l-酒石酸(iiia),随后用156.0g水冲洗。将悬浮液在1小时内加热至内部温度为75℃,然后在75℃下搅拌3小时。随后,使用冷却斜坡,将混合物在5.0小时内冷却至23℃,然后在该温度下搅拌过夜(约18小时)。将悬浮液通过玻璃料滤出,并用由178g酒精和75g水组成的混合物洗涤两次。湿产量:255.4g。然后在50℃下在减压(<100毫巴)下将湿产物干燥过夜(约16小时)。产量:188.6g(理论值的100.73%)无色晶体粉末。将母液和洗涤液合并(约3200ml淡黄色溶液,ph=4.6),并从中分离粗制芬尼酮(i)(实施例7b)。以类似于实施例1b和1c中所述的方式,芬尼酮(ia)的对映体可由该酒石酸盐制备。实施例7b从母液中分离粗芬尼酮(i)在室温下,通过加入43.3g磷酸钠水溶液(100g溶于1l水中)将实施例7a中合并的母液和洗涤液(约3200ml淡黄色溶液,ph=4.6)调节至ph=7.6。然后在减压(85至65毫巴,内部温度38至20℃)下,将酒精基本上蒸馏出,并将混合物减少至最终体积为约0.8l。将混合物冷却至室温,并且在22℃下将沉淀的悬浮液搅拌2小时。抽滤悬浮液,并且以每次150ml水洗涤两次。湿产量:159.1g。在50℃下在减压(<100毫巴)下将湿产物干燥过夜(约16小时)。产量:86.3g(基于实施例7a中所用的外消旋体(ii)计,理论值的92.2%)。以类似的方式,如实施例1c中所述,该粗产物可用于制备纯的形式的芬尼酮(i)。实施例8实施例8a从母液中分离错误的对映体(wrongenantiomer)(ia)在室温下,通过添加101.1g磷酸钠水溶液(100g溶于1l水中)将实施例1a中合并的母液和洗涤液(约3750ml淡黄色溶液,ph=4.5)调节至ph=7.5。然后在减压(85至65毫巴,内部温度38至20℃)下,将酒精基本上蒸馏出,并将混合物减少至最终体积为约0.85l。将混合物冷却至室温,并将沉淀的悬浮液搅拌整个周末(>48小时),然后在22℃下再搅拌2小时。抽滤悬浮液,并且以每次200ml水洗涤两次。湿产量:139.1g。在50℃下在减压(<100毫巴)下将湿产物干燥过夜(约16小时)。产量:103.1g(基于实施例1a中所用的外消旋体(ii)计,理论值的82.48%)。实施例8b用于分离出芬尼酮对映体(ia)的工业批料对于来自实施例4(工业规模的酒石酸盐制备)的合并的母液和洗涤液进行的再处理如下:溶液的量约1165kg·磷酸钠溶液的制备:在20℃下,将7.3kg磷酸钠溶解于73.1kg水中。·在1600l搅拌式容器中,在20℃下首先加入1165kg母液(包括洗涤液),并使用28kg上述制备的磷酸钠溶液将其调节至ph7.5(+/-0.1)。·将混合物搅拌0.25小时,随后在减压(65毫巴,it约22℃)下蒸馏出酒精至装料高度为约310l。·然后在20℃下将混合物搅拌2小时,并使用翻袋式离心机分离悬浮液,每次以20kg水洗涤。·然后在50℃下、在减压(30毫巴;约6小时)下、在球式干燥器中,将湿产物干燥。·将混合物冷却至15℃,并将产物移出。批料1使用的母液(包括洗涤液)的分析数据批料2使用的母液(包括洗涤液)的分析数据批料投入kg产量:kg收率:理论值的%批料1~1165的母液(包括洗涤液)38.296批料2~1165的母液(包括洗涤液)37.193分析数据批料1芬尼酮对映体(ia)纯度:99.71%对映体过量96.56%e.e.最大量的次级组分杂质e0.106%(+)-o,o-二苯甲酰基-d-酒石酸0.081%分析数据批料2芬尼酮对映体(ia)纯度:99.446%对映体过量99.5%e.e.最大量的次级组分杂质e0.107%(+)-o,o-二苯甲酰基-d-酒石酸0.33%实施例9其他酒石酸衍生物和其他溶剂的实施例实施例9a(+)-o,o-二-对茴香酰基(p-anisoyl)-d-酒石酸将100mg外消旋体和111mg(+)-o,o-二-对茴香酰基-d-酒石酸溶解于5ml二氯甲烷中并静置。一段时间后,沉淀出非对映体盐。将盐滤出并且测定对映体过量。测量显示(ia)的对映体过量为68%e.e.。实施例9b(-)-o,o′-二-对甲苯甲酰基(p-toluyl)-l-酒石酸将100mg外消旋体和0.55当量的(-)-o,o′-二-对甲苯甲酰基-l-酒石酸溶解于4ml溶剂中并静置。一段时间后,沉淀出非对映体盐。将盐滤出并且测定对映体过量。测量显示(ia)的对映体过量为y%e.e.。结果总结于下表7中。表7:4ml溶剂/溶剂混合物对映体过量e.e.%(ia)正丙醇/水80∶2085.35正戊醇/水90∶1089.10甲醇/水80∶2084.47甲醇93.18异丙醇/水80∶2081.20异丁醇/水90∶1079.25乙二醇/水80∶2093.68乙二醇68.50乙醇/水80∶2085.87乙醇85.962-丁醇/水80∶2083.921-丁醇/水90∶1088.93实施例9c(+)-o,o-二苯甲酰基-d-酒石酸将100mg外消旋体和0.55当量的(+)-o,o-二苯甲酰基-d-酒石酸溶解于4ml溶剂中并静置。一段时间后,沉淀出非对映体盐。将盐滤出并且测定对映体过量。测量显示(i)的对映体过量为y%e.e.。结果总结于下表8中。表8:4ml溶剂/溶剂混合物对映体过量e.e.%(i)正丙醇/水80∶2094.01正戊醇/水90∶1094.59甲醇/水80∶2089.09甲醇91.21异丙醇/水80∶2090.92异丁醇/水90∶1085.42乙二醇92.08乙醇/水80∶2094.64乙醇93.64环己醇/水90∶1084.33苯甲醇/水90∶1088.832-丁醇/水80∶2092.491-戊醇80.991-丁醇/水90∶1094.02附图说明图1:根据实施例1a以无色晶体粉末形式获得的化合物(vi)的xrpd。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1