用于细胞谱系的多重定量分析的组合物和方法
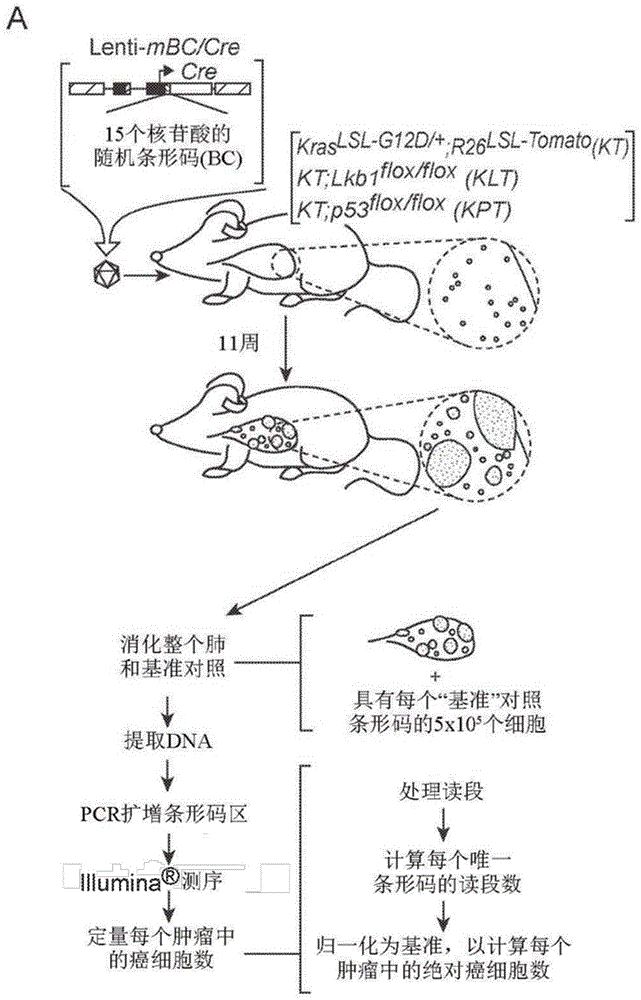
关于联邦政府赞助研究的声明本发明是受美国政府的支持按国立卫生研究所/国家癌症研究所的合同号r01ca207133进行的。相关申请的交叉引用本申请要求2018年10月2日提交的美国临时申请号62/740,311的权益,该申请以引用的方式整体并入本文。
背景技术:
:基因组测序已在全基因组水平上对人癌症中的体细胞改变进行了编目,并且己鉴定出很多潜在重要的基因(例如,推定的肿瘤抑制基因、推定的癌基因、可以引起治疗耐受性或敏感性的基因)。然而,基因组改变的鉴定不一定表示这些改变在癌症中具有功能重要性,并且基因失活或改变的影响单独或与其他遗传改变(体细胞改变或种系改变)或微环境差异组合仍然难以从单独癌症基因组测序数据中被收集。技术实现要素:已经在细胞系以及基因工程小鼠模型系统中使用敲低、敲除和过表达研究来直接研究了遗传改变对赘生物生长的分子和细胞影响。在过去几十年中,培养中的癌细胞系中的基因功能分析已经提供了对癌症的很多方面的见解。然而,培养中的癌细胞系的接近最佳生长、广泛预先存在的遗传和表观遗传变化、以及自身微环境的缺乏限制了这些系统提供对不同的基因如何推动或驱动体内表型(例如,癌症生长、转移、疗法响应)的见解的能力。相比之下,人癌症的基因工程小鼠模型有利于将确定的遗传改变引入正常成体细胞中,这导致了肿瘤在其天然体内环境中的启动和生长。这是特别重要的,因为很多途径受到体内肿瘤微环境的性质的影响。虽然体内系统(诸如基于crispr/cas的遗传靶向)已经扩大了体外和体内功能分析的规模,但是体内系统仍然继续依赖于相对粗略的肿瘤生长的测量,这限制了它们在具有最显著作用的基因分析中的应用。用于分析体内基因功能的严格定量系统的缺乏阻止了对驱动或驱使肿瘤生长、或影响致癌作用的任何其他重要方面的途径(例如,肿瘤抑制途径)的广泛理解。需要有利于在具有多种克隆细胞群(例如,多种可区分的细胞谱系—它们是不同的、可识别的肿瘤,或肿瘤内的不同的、可识别的亚克隆)的个体中进行克隆群体大小(例如,每个肿瘤的大小、每个肿瘤或亚克隆中的赘生性细胞的数量等等)的精确定量的组合物和方法。本公开的组合物和方法满足了该需求,并且提供了揭示不同的单独基因(例如,肿瘤抑制物、癌基因)或遗传改变(例如,插入、缺失、点突变)或者基因和/或遗传改变的组合,是否对细胞群体生长(例如,肿瘤生长)以及其他重要表型(例如,肿瘤进化、进展、转移倾向)具有不同的总体作用的能力。本公开的组合物和方法还提供了测试潜在疗法(例如,放疗、化疗、禁食、化合物(诸如药物)、生物制剂等)对相同的组织内(例如,相同的个体内)的多个不同的克隆细胞群(例如,具有相似的基因型但是具有不同的启动事件的多种肿瘤,具有不同的基因型的多种肿瘤等等)的生长的作用的能力,这将大大减少由样品间差异(例如,动物间差异)所引入的误差。这些方法还有利于合理的药物组合的开发和测试。提供了用于测量相同的组织中(例如,相同的个体中)或不同的组织中的多个克隆细胞群的群体大小的组合物和方法。作为一个实例,在一些情况下,主题方法是测量相同的组织中(例如,相同的个体中)的多个克隆独立的肿瘤细胞群(例如,不同的肿瘤)的肿瘤大小的方法。作为一个示例性实例,如下文在工作实例中所述,本发明人将细胞条形码编制(barcoding)(例如,肿瘤条形码编制)和高通量测序(在工作实例中称为“tuba-seq”)与人癌症的基因工程小鼠模型组合,从而以前所未有的分辨率来定量肿瘤生长。单独的肿瘤大小的精确定量使他们发现使不同的肿瘤抑制基因(例如,已知的肿瘤抑制基因)失活的影响。此外,本发明人将这些方法与多重crispr/cas9介导的基因组编辑整合,这能够对一组推定的肿瘤抑制基因进行平行失活和功能定量–从而对功能性肺部肿瘤抑制物进行鉴定。该方法是研究遗传改变对体内癌症生长的影响的快速、多重和高度定量平台。另外如以下工作实例中所述,本发明人将多重体细胞同源定向修复(hdr)与具有条形码的hdr供体模板一起使用,在单独的小鼠内产生了具有遗传多样性的条形码的肿瘤(例如,在确定的基因中具有遗传多样性的点突变的肿瘤),并且采用定量肿瘤分析(使用高通量测序)来同时在相同的动物中快速和定量查询多个精确突变(例如,确定的点突变)的功能。在一些实施方案中,主题方法包括使组织(例如,肌肉、肺、支气管、胰腺、乳腺、肝、胆管、胆囊、肾、脾、血液、肠、脑、骨、膀胱、前列腺、卵巢、眼、鼻、舌、口、咽、喉、甲状腺、脂肪、食管、胃、小肠、结肠、直肠、肾上腺、软组织、平滑肌、脉管系统、软骨、淋巴、前列腺、心脏、皮肤、视网膜以及繁殖和生殖系统(例如,睾丸、繁殖组织)等)与可遗传且可彼此区分的多种细胞标志物接触的步骤,以在所接触的组织中产生可遗传标记的细胞的多种可区分的谱系。在一些实施方案中,用于接触组织的细胞标志物是具有条形码的核酸(例如,rna分子;或环状或线性dna分子(诸如质粒、天然或合成的单链或双链核酸片段和小环))。在一些实施方案中(例如,在其中细胞标志物是具有条形码的核酸的情况下),细胞标志物可以经由病毒载体(例如,慢病毒载体、腺病毒载体、腺相关病毒(aav)载体和逆转录病毒载体)递送至组织。在一些情况下,待接触的组织已经包括与细胞标志物接触之前的赘生性细胞。在一些情况下,细胞标志物可以诱导赘生性细胞形成和/或肿瘤形成。在一些情况下,与细胞标志物相连接的组分可以诱导赘生性细胞形成和/或肿瘤形成。在一些情况下,细胞标志物是可以诱导赘生性细胞形成和/或肿瘤形成的具有条形码的核酸(例如,同源定向修复(hdr)dna供体模板;编码一种或多种基因组编辑蛋白的核酸;编码癌基因的核酸;编码一种或多种蛋白的核酸,例如一种或多种野生型和/或突变型蛋白[例如,编码对肿瘤有害(以除生长/增殖之外的某种方式)的蛋白的野生型或突变型cdna];crispr/cas指导rna;短发夹rna(shrna);编码针对其他基因组编辑系统的靶向组分的核酸;等)。主题方法还可以包括(在对于至少一部分可遗传标记的细胞经历至少一轮分裂而言已过去足够的时间之后)检测和测量所接触的组织中存在的多种细胞标志物中的至少两种的数量-从而产生一组测量值的步骤,所述测量值表示保留在所接触的组织中的细胞标志物(例如,与所标记的细胞可遗传相关)的身份和数量。在一些情况下(例如,当细胞标志物是具有条形码的核酸时),检测和测量可以经由包括对每个所检测的条形码的序列读段的高通量测序以及对所述读段的数量的定量的方法来进行。在一些情况下,所产生的一组测量值被用作输入来计算(例如,使用计算机)所接触的组织中存在的可遗传标记的细胞数(例如,对于至少2种、至少3种、至少4种、至少5种、至少100种、至少1000种、至少10000种或至少100000种可遗传标记的细胞的所检测的可区分的谱系)(例如,在一些情况下在10至1000000种、10至100000种、10至10000种、或10至1000种可遗传标记的细胞的所检测的可区分的谱系的范围内)。所计算的可遗传标记的细胞数可以是绝对的(例如,确定存在的实际细胞数),或可以是相对的(例如,可以相对于可遗传标记的细胞的第二谱系的群体大小确定可遗传标记的细胞的第一谱系的群体大小,而不必确定任一谱系中存在的实际细胞数)。在一些实施方案中,主题方法包括将测试化合物(例如,药物)施用于组织(例如,经由向个体的施用,经由接触合成的离体组织(诸如类器官)等等)的步骤,例如在引入细胞标志物之后,例如在经由使组织与多种细胞标志物接触来诱导赘生性细胞(或亚克隆)的步骤之后。在一些此类情况下,施用测试化合物的步骤之后是测量多个标记的细胞谱系/细胞群的群体大小(例如,肿瘤大小、每个肿瘤中的赘生性细胞的数量)的步骤。因为可以测量对于相同的组织内(例如,相同的动物内)的独特的和可区分的标记细胞谱系的多个细胞群(例如,可以测量多个肿瘤大小),由于药物响应的样品间差异(例如,动物间差异)而产生的误差的风险如果不能消除,也可以大大减小。在一些方面,本公开提供了一种测试治疗对多个克隆细胞群的效果的方法,所述方法包括:(a)使组织与核酸细胞标志物接触以产生标记的细胞;(b)使所述组织中的所述标记的细胞生长以产生具有可区分谱系的可遗传标记的克隆细胞群;(c)使所述组织中的所述克隆细胞群经受疗法;并且(d)测量所述组织中具有可区分谱系的可遗传标记的细胞。在一些实施方案中,所述细胞标志物用选自由慢病毒载体、腺病毒载体、腺相关病毒载体、逆转录病毒载体、博卡病毒载体和泡沫病毒载体组成的组的病毒载体递送。在一些实施方案中,所述细胞标志物是病毒编码的唯一dna序列。在一些实施方案中,所述细胞标志物包含具有附于其表达的阅读框的3'末端的唯一rna序列的病毒编码的可表达基因。在一些实施方案中,所述细胞标志物还包含任选具有激活突变的肿瘤促进基因。在一些实施方案中,所述细胞标志物还包含靶向所关注的基因的grna,所述所关注的基因任选地是肿瘤抑制物。在一些实施方案中,所述细胞标志物包含多个肿瘤促进基因,并且其中所述细胞标志物包含标识所述肿瘤促进基因的条形码。在一些实施方案中,所述细胞标志物包含多个肿瘤促进基因,其中所述细胞标志物包括:a)标识所述肿瘤促进基因的多核苷酸条形码序列,和b)标识所述单个核酸和从所述单个核酸生长的克隆物的多核苷酸唯一分子标识符(umi)序列。在一些实施方案中,所述组织在动物体内,并且所述疗法全身性施用。在一些实施方案中,所述组织在动物体内,并且所述疗法以组织特异性方式施用。在一些实施方案中,所述疗法选自由小分子、放射、化学疗法、禁食、抗体、免疫细胞疗法、酶、病毒和生物制剂组成的组。在一些实施方案中,所述测量包括从所述组织中分离核酸,扩增所述细胞标志物,并通过测序来定量所述细胞标志物。在一些方面,本公开提供了一种核酸,所述核酸从5′至3′包含:(a)rna聚合酶iii启动子,所述启动子包含由终止密码子隔开的两个杂交tata/frt序列,(b)编码rna的开放阅读框,和(c)泛染色质开放元件(ucoe);其中所述启动子与所述编码rna基因的开放阅读框可操作地连接,并且ucoe与所述rna聚合酶iii启动子可操作地连接,并且其中在通过翻转酶(flp)进行重组后,所述rna的表达得以激活。在一些实施方案中,所述rna聚合酶iii启动子是3型启动子rna聚合酶iii启动子或是来自酿酒酵母(saccharomycescerevisiae)的u6rna启动子。在一些实施方案中,杂交tata/frt序列是seqidno:8(5′-gaagttcctattctctataaagtataggaacttc-3′)。在一些实施方案中,所述ucoe源自异染色质蛋白的无甲基化岛。在一些实施方案中,所述ucoe源自cbx1的无甲基化岛。在一些实施方案中,所述ucoe是seqidno:9。在一些实施方案中,所述核酸还包含条形码以标识rna基因。在一些实施方案中,所述rna是crispr指导rna(grna)。在一些实施方案中,所述核酸还包含编码cre重组酶的基因。在一些方面,本公开提供了一种用于产生具有第一所关注的基因的敲除以及第二所关注的基因的条件性crispr靶向的细胞的系统,所述系统包括:(a)真核细胞,所述真核细胞包含:(i)通过由第一重组酶靶向的重组位点侧接在其5'末端和3’末端上的所述所关注的基因和(ii)在配体诱导型系统的控制下的翻转酶(flp)重组酶;(b)病毒载体,所述病毒载体包含权利要求67所述的核酸,还包含第一重组酶,其中所述rna是针对第二基因的grna;其中在所述真核细胞与所述病毒载体接触时,所述第一所关注的基因失活,并且其中在施用所述配体时,所述grna的表达被激活以裂解在所述第二所关注的基因内的序列。在一些实施方案中,所述配体诱导型系统是雌激素受体(er)与flp的融合。在一些实施方案中,所述第一重组酶是cre、dre、φc31整合酶、kd酵母重组酶、r酵母重组酶、b2酵母重组酶或b3酵母重组酶。在一些实施方案中,所述第一重组酶是cre并且所述重组位点是loxp位点。在一些实施方案中,所述病毒载体是慢病毒载体、腺病毒载体、腺相关病毒载体、逆转录病毒载体、博卡病毒载体或泡沫病毒载体。在一些实施方案中,所述配体诱导型系统是在四环素诱导型启动子、他莫西芬诱导型启动子、蜕皮激素诱导型启动子或孕酮诱导型启动子的控制下的flp。在一些实施方案中,所述系统包含具有多个不同的grna序列的多个病毒载体。在一些实施方案中,所述系统包含具有多个不同的grna序列的多种病毒载体,其中所述多个不同的grna序列针对所述组织内源的多个基因。在一些实施方案中,所述系统包含具有多个不同的grna序列的多种病毒载体,其中所述多个不同的grna序列针对所述组织内源的单个基因。在一些方面,本公开提供了一种含有多个克隆细胞群的动物,其中所述多个克隆细胞群还包含具有从与细胞标志物接触的组织生长的具有可区分谱系的带可遗传条形码的细胞。在一些实施方案中,所述多个克隆细胞群是至少5、10、50、100、200或500个细胞群。在一些实施方案中,所述克隆细胞群包含多种不同的致癌基因组改变。在一些实施方案中,所述遗传条形码包括标识所述单个致癌基因组改变的唯一序列。在一些实施方案中,所述遗传条形码包括标识与所述组织接触的细胞标志物的单个分子的唯一分子标识符序列(umi)。在一些实施方案中,所述遗传条形码是基因组dna中的非转录序列。在一些实施方案中,所述遗传条形码处于连同所述细胞标志物一起引入所述细胞中的可表达基因的转录部分内。在一些实施方案中,所述多个致癌基因组改变包括癌基因中的至少一个激活突变。在一些实施方案中,所述激活突变处于内源性癌基因中。在一些实施方案中,在靶向所述内源性癌基因的sgrna旁边引入所述激活突变。在一些实施方案中,sgrna靶向内源性癌基因的内含子。在一些实施方案中,内源性癌基因是kras,并且sgrna靶向kras的内含子2。在一些实施方案中,所述激活突变处于是癌基因的转基因中。在一些实施方案中,所述多个基因组改变包括肿瘤抑制基因中的至少一个失活基因改变。在一些实施方案中,所述肿瘤抑制基因中的所述失活基因改变至少是功能所需的所述基因或所述基因的一部分的切除。在一些实施方案中,所述肿瘤抑制基因中的所述失活基因改变至少是消除所述基因的转录或引起导致所述基因提前终止的移码突变的插入缺失。在一些实施方案中,所述多个基因组改变包括癌基因中的至少一个激活突变和肿瘤抑制基因中的至少一个失活基因改变。在一些实施方案中,所述多个致癌基因组改变包括癌基因中的多个激活突变和多个肿瘤抑制基因中的至少一个失活基因改变。在一些实施方案中,所述癌基因至少是hras、kras、pik3ca、pik3cb、egfr、pdgfr、vegfr2、her2、src、syk、abl、raf或myc。在一些实施方案中,所引入的激活突变经由引入到所述癌基因的至少3、5、8或10个密码子的摆动碱基中的条形码标识,或者经由引入到所述癌基因的内含子中的条形码标识。在一些实施方案中,所述肿瘤抑制基因是p53、lkb1、setd2、rb1、pten、nf1、nf2、tsc1、rnf43、ptprd、fbxw7、fat1、lrp1b、rasa1、lats1、arhgap35、ncoa6、ncor1、smad4、keap、ubr5、mga、clc、atf7ip、gata3、rbm10、cmtr2、arid1a、arid1b、arid2、smarca4、dnmt3、tet2、kdm6a、kmt2c、kmt2d、dot1l、ep300、atrx、brca2、bap1、ercc4、pole、atm、wm、cdkn2a、cdkn2c或stag2。在一些实施方案中,所述细胞的基因组还包含靶向肿瘤抑制基因的指导rna。在一些实施方案中,所述细胞的基因组还包含:(a)标识所述指导rna的条形码序列和(b)标识所述细胞标志物分子的唯一分子标识符(umi)序列。在一些实施方案中,所述细胞的基因组包含侧接所述肿瘤抑制基因或其关键片段的重组酶位点,并且所述致癌改变至少是重组酶介导的所述肿瘤抑制基因或其关键片段的切除。在一些实施方案中,所述细胞包含带有激活突变的癌基因转基因,所述激活突变具有侧翼是癌基因orf的5’的重组酶位点的终止密码子,从而防止所述转基因的转录,并且所述致癌改变至少是通过所述重组酶切除所述密码子,从而激活所述转基因的表达。在一些实施方案中,重组酶位点是cre、dre、φc31整合酶、kd酵母重组酶、r酵母重组酶、b2酵母重组酶或b3酵母重组酶的重组酶位点。通过引用并入本说明书中提到的所有出版物、专利和专利申请案均通过援引并入本文,其程度如同特别且单独地指出将每个单独的出版物、专利或专利申请通过援引并入一样。附图说明本发明的新颖特征在所附权利要求书中详细阐述。通过参考以下详细描述,将获得对本发明特征和优点的更好理解,以下详细描述阐述了说明性实施方案,其中利用了本发明的原理。结合附图阅读以下详细描述时本发明得到最佳理解。应该强调,根据通常实践,附图的各种特征结构未按比例绘制。相反,为清晰起见,各种特征结构的尺寸可随意放大或缩小。附图包括以下图解。图1.tuba-seq将肿瘤条形码编制与高通量测序组合在一起,可以对肿瘤大小进行平行定量。a,评估肺部肿瘤大小分布的tuba-seq流程(pipeline)的示意图。在kraslsl-g12d/+;rosa26lsl-tomato(kt)、kt;lkb1flox/flox(klt)和kt;p53flox/flox(kpt)小鼠中用lenti-mbc/cre(含有随机的15个核苷酸的dna条形码(bc)的病毒)来启动肿瘤。经由来自具有肿瘤的肺的dna的成批条形码测序来计算肿瘤大小。b,具有lenti-mbc/cre启动的肿瘤的kt、klt和kpt小鼠的肺叶的荧光解剖镜图像。肺叶的轮廓以白色虚线勾勒。显示了lenti-mbc/cre的滴度。在不同的遗传背景中使用不同的滴度来产生大致相等的总肿瘤负荷,尽管总体肿瘤生长存在差异。上图中的比例尺=5mm。下图中的比例尺=1mm。c,kt、klt和kpt小鼠中的肿瘤大小分布(显示了每组的小鼠数量)。每个点表示一个肿瘤。在每个肿瘤中,每个点的面积与癌细胞的数量成比例。1mm直径的球形肿瘤中的癌细胞的大致数量所对应的点在数据的右侧显示,以供参考。图2.tuba-seq是定量肿瘤大小的稳健的和可重现的方法。a,dada2是被设计用于扩增子数据的深度测序的去噪算法,它消除了可能表现为假性肿瘤的反复出现的读段误差。将具有已知条形码的细胞系添加至来自每只小鼠的每份肺部样品(每份样品5x105个细胞)。来源于这些已知条形码的反复出现的读段误差表现为约5000个细胞的假性肿瘤。dada2鉴定并大大减少了这些反复出现的读段(测序)误差。b,c,从单独的成批肺部样品制备的技术重复测序文库展示出单独的病灶大小(b)和大小谱(c)(显示了第50个至第99.9个百分位处的肿瘤)之间的高度对应性。d,我们的分析流程对于读段深度的差异、dna条形码的gc含量、以及条形码文库的多样性而言是稳健的。将肿瘤分为对应于每个技术参数的高、中和低水平的三份:测序深度、肿瘤条形码的gc含量、以及唯一条形码的估算数量(参见方法)。箱须帽为1.5iqr。e,五只klt小鼠中的大小分布的再现性。虽然肿瘤大小的小鼠间差异较小,但是小鼠具有总体相似的大小谱。单独的小鼠中的指定百分位处的肿瘤大小通过直线来连接。f,当比较相同小鼠内的肿瘤时,大小谱的再现性得到改善,这表明肿瘤大小的小鼠间差异较大。将每只小鼠中的肿瘤分成两组,并比较这些组的大小谱。单独的小鼠中的指定百分位处的肿瘤大小通过直线来连接。g,基于指定百分位处的肿瘤大小之间的总最小二乘距离的kt、kpt和klt小鼠的无监督层次聚类(通过ward方差最小化算法进行聚类)。根据基因型进行的小鼠聚类表明,tuba-seq识别了每种基因型的大小谱的可重现差异。图3.肿瘤大小的大规模平行定量使多个基因型之间的分布拟合成为可能。a,b,klt(n=5)小鼠(a)和kpt(n=3)小鼠(b)中的指定百分位处的肿瘤大小与kt小鼠(n=7)中的指定百分位处的肿瘤大小。使用利用lenti-mbc/cre进行肿瘤启动后11周的每种基因型的全部小鼠的全部肿瘤来计算每个百分位数。c,相对于相同百分位处的kt肿瘤而言,对于每种基因型的指定百分位处的肿瘤大小。误差条是经由自举获得的95%置信区间。与对应的kt百分位显著不同的百分位以彩色表示。d,正如对于具有正态分布生长速率的指数肿瘤生长所预期的那样,肿瘤大小分布与对数正态分布的拟合最密切。在整个完整的肿瘤大小谱中,对数正态分布最好地描述了klt小鼠中的肿瘤(中图)。通过将较小尺度的对数正态分布与较大尺度的幂律分布组合能够很好地解释kt小鼠(左图)和kpt小鼠(右图)中的肿瘤大小分布。在考虑单独的基因(或基因的组合)如何导致肿瘤生长加快时,这些差异是至关重要的。幂律关系在对数-对数轴上线性下降,这与kt小鼠中的前约1%的肿瘤和kpt小鼠中的约10%的肿瘤中的罕见、但非常大的肿瘤一致。注意:在11周后仅kpt小鼠中的肿瘤还超过一百万个细胞,这与我们的研究中能够产生最大的肿瘤的p53缺陷一致。图4.使用tuba-seq和多重crispr/cas9介导的基因失活进行的肿瘤抑制表型的快速定量。a,含有双组分条形码的lenti-sgts-pool/cre载体的示意图,所述双组分条形码具有连接至每个sgrna的8个核苷酸的“sgid”序列以及随机的15个核苷酸的随机条形码(bc)。b,lenti-sgts-pool/cre含有四个具有无活性sgrna的载体和十一个靶向已知的和候选肿瘤抑制基因的载体。每个sgrna载体含有唯一的sgid和随机条形码。nt=非靶向。c,多重crispr/cas9介导的肿瘤抑制物失活与tuba-seq结合以评估每个靶基因对肺部肿瘤生长的体内功能的示意图。用lenti-sgts-pool/cre病毒在kt和kt;h11lsl-cas9(kt;cas9)小鼠中启动肿瘤。d,在用lenti-sgts-pool/cre进行肿瘤启动后12周的kt和kt;cas9小鼠的肺叶的明视野(上图)和荧光解剖镜图像(下图)。在荧光图像中,肺叶的轮廓以白色虚线勾勒。显示了病毒滴度。比例尺=5mm。e,组织学确认,kt小鼠具有增生和小肿瘤,而kt;cas9小鼠具有更大的肿瘤。显示了病毒滴度。上图比例尺=3mm。下图比例尺=500μm。图5.tuba-seq以前所未有的分辨率发现了已知的和新型肿瘤抑制物。a,在用lenti-sgts-pool/cre进行肿瘤启动后12周,kt;cas9小鼠中的相对肿瘤大小的分析鉴定出六个肿瘤生长抑制基因。指定百分位处的相对肿瘤大小表示来自8只小鼠的合并数据,这些数据已归一化为sginert肿瘤的平均大小。显示了95%置信区间。显著大于sginert的百分位以彩色表示。b,假设肿瘤大小呈对数正态分布,平均肿瘤大小的估算值鉴定出使kt;cas9小鼠中的生长显著增加的sgrna。显示了邦费罗尼(bonferroni)校正的自举p值。p值<0.05,并且它们对应的平均值以粗体表示。c,针对肿瘤启动后12周的kt和kt;cas9小鼠和肿瘤启动后15周的kt;cas9小鼠中具有每个sgrna的肿瘤的第95个百分位的肿瘤相对大小(左图)、对数正态(ln)平均值(中图)和对数正态(ln)p值(右图)。d,kt;cas9小鼠相对于kt小鼠的总sgid呈现的倍数变化(δsgid呈现)鉴定出若干增加呈现的sgrna,这与被靶向的肿瘤抑制基因失活产生的肿瘤生长增加一致。δsgid呈现是具有kt;cas9小鼠与kt小鼠中的每种sgid的读段的百分比的倍数变化,该倍数变化被归一化为对于sginert,δsgid呈现=1。显示了平均值和95%置信区间。e,f,与仅并入sgrna呈现相比,通过并入来自条形码测序的单独的肿瘤大小可以显著提高检测肿瘤抑制作用的能力。所有当前方法都依赖于sgrna呈现,这远不及tuba-seq。第95个百分位的肿瘤相对大小和通过tuba-seq确定的对数正态统计学显著性与δsgid呈现的平均倍数变化及其相关的p值相比鉴定出更多的作为肿瘤抑制物的基因(e和f)。(e)中的误差条是95%置信区间。(f)中的点线表示0.05的显著性阈值。点的颜色对应于图4b中sgrna的颜色。图6.独立的方法将setd2鉴定为肺部肿瘤生长的有效抑制物。a,靶基因座处的含有插入缺失的读段的百分比被归一化为含有3个独立的新霉素基因座中的插入缺失的读段的平均百分比。对于三只单独的小鼠,将该值对针对每个sgrna的第95个百分位的肿瘤大小进行作图。我们展示了setd2、lkb1和rb1中的高频率插入缺失与针对中靶sgrna切割的选择一致。每个点表示来自单只小鼠的sgrna。根据图4b,sgneo点以黑色表示,所有其他点以彩色表示。b,在肿瘤启动后9周分析的来自用lenti-sgsetd2#1/cre、lenti-sgsetd2#2/cre或lenti-sgneo2/cre感染(转导)的kt;cas9小鼠的肺叶的荧光解剖镜图像和h&e。在荧光解剖镜图像中,肺叶的轮廓以白色虚线勾勒。上图比例尺=5mm。下图比例尺=2mm。c,通过组织学进行的肿瘤面积百分比的定量显示,与用lenti-sgsetd2#1/cre或lenti-sgsetd2#2/cre感染(转导)的kt小鼠相比,相同病毒感染(转导)的kt;cas9小鼠中的肿瘤负荷显著增加。每个点表示小鼠,条是平均值。*p值<0.05。ns=不显著。d,来自具有lenti-sgsetd2#1/cre启动的肿瘤与lenti-sgneo2/cre启动的肿瘤的kt;cas9小鼠的指定百分位处的肿瘤大小(n=4只小鼠/组)。使用来自每组中的所有小鼠的所有肿瘤来计算百分位。图7.人肺部腺癌中的基因组改变的频率以及肿瘤启动和条形码编制的描述。a,显示了对于所有肿瘤(所有)以及具有致癌性kras突变(krasmut)的肿瘤中在每个肿瘤抑制基因中具有潜在失活的改变(移码突变或非同义突变,或者基因组丧失)的肿瘤的百分比。显示了每个数据集中kras中具有致癌性突变的肿瘤的数量和百分比。b,已编制条形码的慢病毒-cre载体的吸入在基因工程小鼠模型中启动了肺部肿瘤。重要的是,慢病毒载体稳定整合进所转导的细胞的基因组中。可以通过基于高通量测序的方法来确定每个具有唯一条形码的细胞的相对扩增。c,来自用1.7x104个lenti-cre病毒感染(转导)的kraslsl-g12d/+;r26lsl-tomato(kt)小鼠的肺部组织切片的苏木精和伊红(h&e)染色。这些小鼠产生了赘生性细胞的小扩增以及较大的腺瘤。比例尺=50μm。图8.体内定量肿瘤大小的tuba-seq流程。a,经整合的慢病毒载体的dna条形码区的测序能够精确测量病灶大小。首先,丢弃具有较差的phred质量得分或不期望的序列的读段。然后,将读段堆积成具有唯一条形码的组。使用dada2从小病灶中划定反复出现的测序误差,dada2是最初被设计为鉴定读段全长的深度测序扩增子的测序误差的模型。通过该聚类算法,将被视为来自大肿瘤的扩增条形码区的反复出现的测序误差的小条形码堆积与这些较大的堆积组合。使用基准对照将读段堆积转换为绝对细胞数。最后,使用测序信息和绝对细胞数二者来建立识别病灶的最小截断值,以使流程的再现性最大化。b,c,唯一的读段堆积可能不对应于唯一的病灶,而是由来自非常大的肿瘤(例如,更大的肿瘤)的条形码的反复出现的测序误差产生。dada2用于合并具有足够大小和序列相似性的较大病灶的小读段堆积。该算法从我们的深度测序区的非简并区(即,侧接条形码的慢病毒载体的区域)来计算测序误差率(b)。针对每个phred得分计算每次转换和颠换(显示了a至c转换和颠换)的可能性,以生成每次运行的特定误差模型(c)。公布的phred误差率(红色)通常低于观察的phred误差率(黑色;用于正则化的loess回归)。然后将这些误差模型(针对每台机器进行训练)用于确定应将较小的读段堆积捆扎成具有强序列相似性(表明所述较小的堆积是反复出现的读段误差)的较大的堆积还是保留为单独的病灶。d-f,我们在三台不同的机器上对第一实验样品(图1的kt、klt和kpt)进行测序,以对dada2进行检查和参数化。预期识别良好病灶的方案显示出(d)在识别病灶的数量方面具有强相似性,(e)在条形码大小之间具有良好相关性,以及(f)在3次运行中具有相似的每个sgid库的平均大小。这三次运行在测序深度(预处理后的40.1x106、22.2x106和34.9x106个读段)方面产生自然变化,并且在每个碱基的预期误差率(0.85%、0.95%和0.25%)方面产生自然变化—这为对方法的一致性进行检查提供了有用的技术干扰。我们发现,在500个细胞处截断病灶大小以及在10-10(红色方块)处截断dada2聚类概率(ω)以非常小的尺度提供了病灶大小的谱分析,同时仍使我们的测试指标的差异最小。图9.基准对照允许计算每种肺部样品中的每个肿瘤中的癌细胞数。a,使用三个具有已知条形码基准对照细胞系的方案的示意图。将每种细胞系的5x105个细胞添加至每份肺部样品。然后从肺部加上全部三个基准对照提取dna,并且对条形码进行pcr扩增和深度测序。然后我们通过以下方式来计算该肺部样品中的每个肿瘤中的癌细胞数:用与基准相关的读段%除以从每个肿瘤观察到的读段%(唯一条形码),并乘以5x105,从而得到癌细胞数。b,两个具有非常不同的肿瘤负荷的肺部的实例。不论总肿瘤负荷为多少都可以使用这些基准细胞系来确定单独的肿瘤中的癌细胞数。还应注意,周围的“正常肺”组织对此计算没有影响,因为该组织没有慢病毒整合,因此将不会有助于读段。基准对照(例如,5x105、5x104、5x103、5x102或50个细胞)的滴定的产生有利于tuba-seq的分辨率扩展至较小的克隆扩增)。图10.基于dada2的肿瘤的识别流程是稳健和可重现的。a,肿瘤大小展示出细微的gc偏差。通过每只小鼠中的每个sgrna的平均大小对每个肿瘤进行的大小对数转换和归一化来使残差肿瘤大小差异最小化。具有中等gc含量的条形码似乎能够最有效地进行pcr扩增。对残差偏差的4阶多项式拟合能够最有效地校正病灶大小。计算该校正并将其应用于所有后续分析,这将使每个病灶大小平均调整5%,并且使每只小鼠中的每个sgid的病灶大小标准偏差相对于平均值仅减少2.9%—这表明虽然gc偏差引入的变化是可测量的,但是该差异是最小的。b,随机条形码在预期的核苷酸之间展示出高度随机性。c,使用tuba-seq获得的所识别的每只小鼠的病灶数量。大于两个不同的细胞数截断值(1000和500)的肿瘤数显示为每只小鼠的肿瘤的平均数±标准偏差。将kt10小鼠暴露于高滴度(6.8x105)(在正文中使用)和低滴度(1.7x105;kt低)。在任一细胞截断值下,对于每个衣壳观察到的肿瘤数量无统计学显著性差异,这表明条形码多样性仍然不限于超过五十万的肿瘤,并且较小的肿瘤不是由肿瘤聚集引起的。d,基于确定百分位处的肿瘤大小之间的总最小二乘距离的kt、kt低、kpt和klt小鼠的无监督层次聚类(通过ward增量算法来确定连接)。具有相同基因型、但是具有不同病毒滴度的小鼠聚类在一起,这表明大小谱差异主要由肿瘤遗传学(基因型),而不是由病毒滴度差异来确定。e,f,病灶大小不受读段深度差异的显著影响。对来自单只小鼠的具有肿瘤的肺的条形码区以非常高的深度进行测序,然后随机下采样至典型的读段深度。对来自单只小鼠的具有肿瘤的肺的条形码区以非常高的深度进行测序,然后随机下采样至典型的读段深度。(e)完全(x轴)和下采样(y轴)数据集的肿瘤大小分布非常相似,这表明读段深度不能使我们的分析参数产生偏差,并且这些分析参数对于读段深度是相当稳健的。(f)在下采样时,百分位计算也是可重现的。g,具有lenti-mbc/cre启动的肿瘤的kt、klt和kpt小鼠(来自图1)具有用六种唯一的lenti-sgid-bc/cre病毒(每种病毒具有唯一的sgid和自然变化的条形码多样性)启动的肿瘤。这允许我们用每只小鼠中的六个重复来定量dada2识别的肿瘤大小差异。当使用来自每只小鼠的所有肿瘤时,以及当使用具有给定sgid的肿瘤的每个子集时,可重复识别肿瘤大小分布。针对kt(左图)、klt(中图)和kpt(右图)绘制指定百分位处的肿瘤大小。每个点表示使用单个sgid中的肿瘤来计算的百分位值。百分位以灰度表示。具有不同sgid的肿瘤大小的六个重复百分位值是难以区分的,因为它们的强相关性意味着针对每个sgid的标志物是高度重叠的。图11.在具有h11lsl-cas9等位基因的小鼠中用慢病毒-sgrna/cre载体启动的肺部肿瘤中的有效基因组编辑。a,使用lenti-sgtomato/cre(lenti-sgtom/cre)病毒载体和h11lsl-cas9等位基因在肺癌模型中测试体细胞基因组编辑的实验的示意图。所有小鼠都是对于r26lsl-tomato等位基因纯合的,以便于确定纯合缺失的频率。b,来自具有lenti-sgtomato/cre启动的肿瘤的kpt;cas9小鼠的肺叶的荧光解剖镜图像。tomato阴性肿瘤的轮廓以虚线勾勒。上图比例尺=5mm;下图比例尺=1mm。c,针对tomato蛋白发现的tomato阳性(pos)、tomato混合(混合)和tomato阴性(neg)肿瘤的免疫组织化学。肿瘤的轮廓以虚线勾勒。比例尺=200μm。d,具有lenti-sgtom/cre启动的肿瘤的四只kpt;cas9小鼠中的tomato表达的定量表明大约一半肿瘤在至少一部分癌细胞中具有靶基因的crispr/cas9介导的纯合失活。显示了tomato阳性、tomato混合和tomato阴性肿瘤的百分比,其中每组中的肿瘤数在括号中显示。e,使用lenti-sglkb1/cre病毒和h11lsl-cas9等位基因在肺中测试体细胞基因组编辑的实验的示意图。f,用lenti-sglkb1/cre感染(转导)的kt和kt;cas9小鼠的肺叶的荧光解剖镜图像显示出kt;cas9小鼠中的肿瘤负荷增加。肺叶的轮廓以白色虚线勾勒。比例尺=2mm。g,相对于kt小鼠,在lenti-sglkb1/cre感染(转导)的kt;cas9小鼠中以肺重量表示的肿瘤负荷增加,这与肿瘤抑制物lkb1的成功缺失一致。正常肺重量以红色点线表示。*p值<0.02。每个点为一只小鼠,条表示平均值。h,显示在kt;cas9小鼠中lenti-sglkb1/cre启动的肿瘤表达cas9蛋白而缺乏lkb1蛋白的蛋白印迹。hsp90显示了上样的蛋白印迹。图12.靶向十一个已知的和候选肿瘤抑制基因的sgrna的选择和表征。a,根据sgrna在每个基因中的位置、它们与剪接接受体/剪接供体(sa/sd)区的接近程度、它们位于注释功能结构域的上游(还是之内)、它们位于所记录的人突变的上游(还是附近)、以及它们的预测中靶切割效率得分(最高得分为1.0;得分越高=活性越大)和脱靶切割得分(最高得分为100.0;得分越高=特异性越大)来选择sgrna(doench等人,naturebiotechnology,2014;hsu等人,2013)。b,来自公开研究的数据汇总,在所述公开研究中这些肿瘤抑制基因在krasg12d驱动的肺癌模型的背景下是失活的。c,每种载体具有唯一的sgid,并且因随机条形码而不同。显示了对于每个载体的sgid和与每个sgrna相关的条形码的估算数。d,评估lenti-sgts-pool/cre中的每个sgrna的初始呈现的实验的示意图。e,如通过来自三次重复感染的样品的测序所测定的lenti-sgts-pool/cre中的每个sgrna的百分比。显示了平均值+/-sd。库中的每个载体的百分比与每个载体的预期呈现仅略有偏离(红色虚线)。图13.体外sgrna切割效率。a,通过用携带每个单独的sgrna的慢病毒感染cas9细胞来评估每个sgrna的体外切割效率的实验的示意图。我们测试了对于每个靶基因座的三个单独的sgrna,并且我们报道了最佳sgrna的切割效率。b,对于每个被靶向的肿瘤抑制物的最佳sgrna的切割效率。通过桑格(sanger)测序和tide分析软件来评估切割效率(brinkman等人,nucl.acidsres.,2014)。c,通过用lenti-sgts-pool/cre感染cas9细胞来评估每个sgrna的体外切割效率的实验的示意图。在感染(转导)后48小时收获细胞,提取基因组dna,pcr扩增14个被靶向的区域,并且对产物进行测序。通过计算每个区的插入缺失的%,并且归一化为库中的呈现和setd2插入缺失%来确定对于库中的每个sgrna的相对切割效率。d,包含无活性neo靶向对照的每个sgrna的相对切割效率。图14.在多个时间点使用tuba-seq进行的肿瘤抑制物的鉴定和验证。a,在肿瘤启动后12周kt小鼠中的每个lenti-sgrna/cre载体的呈现百分比(计算如下:100乘以具有每个sgid的读段数/所有sgid读段数)。因为kt小鼠中不存在cas9介导的基因失活,所以这些小鼠中的每个sgid的百分比表示lenti-sgts-pool/cre库中具有每个sgrna的病毒载体的百分比。b,在用lenti-sgts-pool/cre进行肿瘤启动后12周,kt小鼠(缺乏cas9)中的相对肿瘤大小的分析基本上鉴定出一致的肿瘤大小分布。指定百分位处的相对肿瘤大小表示来自10只小鼠的合并数据,这些数据已归一化为sginert肿瘤的平均值。显示了95%置信区间。与sginert显著不同的百分位以彩色表示。c,假设肿瘤大小呈对数正态分布,平均肿瘤大小的估算值显示了kt小鼠中的预期微小差异。显示了邦费罗尼校正的自举p值。p值<0.05,并且它们对应的平均值以粗体表示。d,在肿瘤启动后12周kt;cas9小鼠中的每个lenti-sgrna/cre载体的呈现百分比(计算如下:100乘以具有每个sgid的读段数/所有sgid读段数)。e,相对于相同百分位处的含有sginert的肿瘤的平均大小而言,对于每个sgrna的指定百分位处的肿瘤大小。显示了用lenti-sgts-pool/cre进行肿瘤启动后15周来自3只kt;cas9小鼠的合并数据。点线表示与inert相比无变化。误差条表示95%置信区间。其中置信区间不与点线重叠的百分位以彩色表示。f,假设肿瘤大小呈对数正态分布,平均肿瘤大小的估算值鉴定出kt;cas9小鼠中具有显著生长优势的sgrna。显示了邦费罗尼校正的自举p值。p值<0.05,并且它们对应的平均估算值以粗体表示。图15.在两个独立的时间点在具有lenti-sgts/cre启动的肿瘤的kt;cas9小鼠中进行的p53介导的肿瘤抑制的鉴定。a,b,在用lenti-sgts-pool/cre进行肿瘤启动后12周(a)和15周(b)kt;cas9小鼠中的相对肿瘤大小的分析在两个时间点使用幂律统计将p53鉴定为肿瘤抑制物。指定百分位处的相对肿瘤大小为分别来自8只和3只小鼠的合并数据,这些数据已归一化为sginert肿瘤的平均值。显示了95%置信区间。显著大于sginert的百分位以彩色表示。显示了幂律p值。请注意,在该实验环境中,仅最大的sgp53启动的肿瘤的大小大于sginert肿瘤的大小。这可能可通过sgp53的相对较低的切割效率来部分解释(图13d)。c-f,通过用具有给定大小的插入缺失的读段数除以具有插入缺失的读段总数来计算p53基因座处的每种大小的插入缺失(从十个核苷酸缺失(-10)至三个核苷酸插入(+3))的百分比。框内插入缺失以灰色显示。我们评估了在感染(转导)后48小时在用lenti-sgts-pool/cre感染(转导)的表达cas9的细胞系中体外产生的p53基因座处的插入缺失谱。(c)对于框外突变无偏好。然后我们分析了疾病进展15周后具有lenti-sgts-pool/cre启动的肿瘤的三只单独的kt;cas9小鼠(d-f)。很少存在与针对扩增的肿瘤中的框外功能丧失改变一致、与p53的肿瘤抑制功能一致的框内插入缺失(-9、-6、-3和+3)。虽然这些分析类型与tuba-seq发现一致,但是这些分析类型相对于tuba-seq平台是不精确的。图16.肿瘤大小分布的分析表明lkb1和setd2缺陷呈对数正态分布。a,b,指定百分位(%ile)处的肿瘤大小,以及sglkb1(a)或sgsetd2(b)于相同百分位处的sginert启动的肿瘤大小。使用所有肿瘤以及来自所有kt;cas9小鼠的每个sgrna来计算每个百分位,其中在肿瘤启动后12周分析lenti-sgts-pool/cre启动的肿瘤(n=8只小鼠)。用虚线表示相对于sginert启动的肿瘤的大小。c,在kt;cas9小鼠中用lenti-sgsetd2/cre启动的肿瘤的概率密度图,lenti-sgts-pool/cre启动的肿瘤显示出对数正态分布的肿瘤大小与klt小鼠中所见的那些肿瘤大小非常类似。这表明setd2缺陷在不显著增加另外的有利改变的产生或对这些改变的耐受性的情况下驱动肿瘤生长。图17.中靶sgrna作用的确认。a,b,通过将具有给定大小的插入缺失的读段数除以每个最前端肿瘤抑制基因内具有插入缺失的读段总数来计算每个插入缺失(从十个核苷酸缺失(-10)至四个核苷酸插入(+4))的百分比。(a)显示出针对setd2、lkb1、rb1的具有lenti-sgts-pool/cre启动的肿瘤的三只kt;cas9小鼠的平均百分比和标准偏差,以及neo中的三个靶位点(neo1-3)的平均值。框内突变以灰色显示。通过对全部三只小鼠和作为单个组的全部三个neo靶位点求平均值来计算针对neo1-3的平均值和标准偏差。通常,很少框内插入缺失(-9、-6、-3和+3)与所扩增的肿瘤中的这些基因中的针对框外功能丧失改变的选择一致。(b)我们还评估了在感染(转导)后48小时在用lenti-sgts-pool/cre感染(转导)的表达cas9的细胞系中体外产生的插入缺失谱。我们未检测到对这些基因组位置中的任一者的框内突变的偏好,这表明kt;cas9小鼠中的偏差最可能是由于具有框外插入缺失(即,无效等位基因)的肿瘤的有利扩增。c,具有lenti-sgsmad4/cre诱导的肿瘤的kt和kt;cas9小鼠的卡普兰-梅尔(kaplan-meier)存活曲线。在存在致癌性krasg12d的情况下,crispr/cas9介导的smad4灭活,不降低存活率,这表明如果有限制,则来自smad4失活的肿瘤生长增加。d,lenti-sgsmad4/cre感染(转导)的kt;cas9小鼠中的大多数肿瘤,与用相同病毒感染(转导)的kt小鼠相比,丧失smad4蛋白表达,这与smad4基因座处的插入缺失产生一致。比例尺=50μm。e,lenti-sgts-pool/cre感染(转导)的kt;cas9小鼠中的若干肿瘤具有明显的乳突组织学、均一的大细胞核,并且是sox9阳性的,这与apc缺陷、kras驱动的肺部肿瘤的已公开表型一致(sanchez-rivera等人,nature,2014)。显示出代表性sox9阴性和sox9阳性肿瘤。比例尺=100μm(上图)和25μm(下图)。图18.显示在具有crispr/cas9介导的setd2失活的小鼠中使用两种单独的sgrna中的每种使肿瘤负荷增加的另外的图像。在肿瘤启动后9周分析的具有用lenti-sgneo2/cre(左图)、lenti-sgsetd2#1/cre(中图)或lenti-sgsetd2#2/cre(右图)启动的肿瘤的kt;cas9小鼠的肺叶的另外的代表性荧光解剖镜图像。肺叶的轮廓以白色虚线勾勒。比例尺=5mm。图19.评估肿瘤抑制基因在肺部腺癌小鼠模型中的功能的系统的比较。显示了肿瘤抑制基因失活(cre/loxp介导的floxed等位基因缺失与crispr/cas9介导的基因组编辑)的方法、通过单独肿瘤的遗传条形码编制来定量肿瘤数量和大小的能力,以及使合并形式的多个基因失活的能力。显示了每个系统的特别相关的优点和缺点,以及示例参考文献。除maresch等人之外,所有突出的研究均在肺癌方面,而maresch等人则使用合并的sgrna转染来研究胰腺癌。使用floxed等位基因来评估肿瘤抑制基因在肺部腺癌模型中的功能的现实通过以下事实最好地示例:在过去15年中,使用floxed等位基因与kraslsl-g12d组合仅研究了我们所查询的肿瘤抑制基因中的六个。定量方法的缺乏还严重阻碍了基因的鉴定,由于已知和未知的技术和生物学变量(例如,肿瘤启动的再现性、性别、年龄和小鼠品系),只鉴定了适度的肿瘤抑制作用。用floxed等位基因来使基因缺失所产生的数据还受到在不同实验室中所用的不同实验设置(例如,不同的病毒滴度、启动后的时间、定量的方法、小鼠品系)之间进行比较的困难的限制。因此,很难从文献中收集不同的肿瘤抑制基因的相对作用。最后,通过肿瘤条形码编制进行的单独肿瘤细胞数的定量不仅提供了前所未有的精度,而且还发现了对肿瘤大小分布的基因特异性作用,该作用可能反映了肿瘤抑制的独特功能机制。图20.小鼠之间的肿瘤大小分布的统计学性质以及sgrna肿瘤大小的协方差。a.在具有lenti-sgpool/cre启动的肿瘤的每只小鼠中每个sgid分布的平均值和方差。小鼠基因型以彩色表示。通常,对于所有基因型,方差随着平均值的平方而增加,这表明病灶大小的对数变换应稳定方差并避免异方差性。一些分布表现出方差的增加大于平均值的平方。b-d.在12周处死的kt;cas9小鼠中查询响应于遗传改变的小鼠间差异。研究了每只小鼠中每个sgrna的lnmle平均值的协方差。小鼠之间的基因型平均大小彼此正相关(例如具有较大的sglkb1肿瘤的小鼠还具有较大的sgsetd2肿瘤。)在全部12个sgrna(合并sginert)中相关矩阵的pca分解发现了可通过单个主成分(pc1)向量解释的相当水平的小鼠间差异。每个点表示映射到pc1上的单只小鼠,这解释了在sgrna平均大小方面观察到75%的小鼠间差异。(b)pc1与肺总重量以及(c)平均病灶大小相关,这表明具有较大的肿瘤的小鼠更易受到强驱动子驱动的肿瘤生长的影响(pc1与sgsetd2和sglkb1大小相关,数据未示出。)(d)当小鼠映射到前两个主成分时,它们不表现出形成独特的聚类。重复小鼠几乎总是相同笼子中饲养的同胞。我们使用主成分混合模型来使外部噪声源最小化(参见方法。)图21.肿瘤进展的数学模型。图22.慢病毒感染(转导)的频率与在相同小鼠中每个病灶及其的最近邻近者之间的大小差异。图23.整合aav/cas9介导的体细胞hdr与肿瘤条形码编制和测序的平台,该平台允许推定致癌性点突变的体内快速引入和功能研究。a-d.定量测量一组确定的点突变的体内致癌性的流程的示意性概览图。产生了aav载体的文库,使得每种aav含有1)对于同源定向修复(hdr)的模板,该模板含有推定的致癌性点突变和在邻近的摇摆碱基中编码的随机dna条形码,2)靶向内源性基因座以进行hdr的sgrna,以及3)激活基因工程小鼠中的条件cas9等位基因(h11lsl-cas9)和其他cre依赖性等位基因的cre重组酶(a)。aav文库被递送至所关注的组织(b)。在转导后,细胞的亚组经历aav/cas9介导的hdr,其中所关注的基因座在sgrna靶位点被cas9切割,并使用aavhdr模板进行修复。这样可以将所期望的点突变和唯一的dna条形码精确地引入靶基因座(c)。如果所引入的突变足以启动肿瘤发生和驱动肿瘤生长,则通过点突变来工程改造的体细胞可以从头产生肿瘤。d,两种独立的方法可以用于分析肿瘤:1)可以对肿瘤进行单独测序以表征靶基因的两个等位基因,或2)可以对来自整个具有肿瘤的成批组织的具有条形码的突变型hdr等位基因进行深度测序,以定量具有每个突变的肿瘤的数量和大小。e.用于cas9介导的hdr进入内源性kras基因座的aav载体库(aav-krashdr/sgkras/cre)。每个载体含有在密码子12和13处具有12个非同义kras突变中的1个(或野生型kras)、pam和sgrna同源区内的沉默突变(pam*)、以及对于单独肿瘤的dna条形码编制的下游密码子的摇摆位置内的8个核苷酸的随机条形码的hdr模板。f.aav-krashdr/sgkras/cre质粒文库中的每个kras密码子12和13等位基因的呈现。g.aav-krashdr/sgkras/cre质粒文库中的条形码区的多样性。图24.aav/cas9介导的体细胞hdr启动可以进展为转移状态的致癌性kras驱动的肺部肿瘤。a.通过aav-krashdr/sgkras/cre的气管内施用来将点突变和dna条形码引入rosa26lsl-tdtomato;h11lsl-cas9(t;h11lsl-cas9)、p53flox/flox;t;h11lsl-cas9(pt;h11lsl-cas9)和lkb1flox/flox;t;h11lsl-cas9(lt;h11lsl-cas9)小鼠中的肺上皮细胞的内源性kras基因座的实验的示意图。b.aav-krashdr/sgkras/cre-处理的lt;h11lsl-cas9、pt;h11lsl-cas9和t;h11lsl-cas9小鼠中的tomato阳性肺肿瘤的代表性图像和组织学。比例尺=5mm。c.用指定的aav载体(含有和不含有sgkras)感染(转导)的小鼠的指定基因型中的肺部肿瘤的定量。每个点代表一只小鼠。用1:10000稀释度的aav-krashdr/sgkras/cre转导的kraslsl-g12d;lt(klt)和kraslsl-g12d;pt(kpt)小鼠产生的肿瘤数为用未稀释的病毒感染(转导)的pt;h11lsl-cas9和lt;h11lsl-cas9小鼠的大约一半。因此,假设aav-krashdr/sgkras/cre文库中的所有krashdr等位基因是致癌性的,这表明aav/cas9介导的hdr在大约0.02%的转导细胞中出现。或者,如果假设aav-krashdr/sgkras/cre文库中只有20%的突变型等位基因驱动肿瘤形成,则hdr的比率为大约0.1%。d.代表性facs图,它显示了具有aav-krashdr/sgkras/cre启动的肺部肿瘤的lt;h11lsl-cas9小鼠胸膜腔内的tomato阳性弥散性肿瘤细胞(dtc)。e.pt;h11lsl-cas9小鼠中来自aav-krashdr/sgkras/cre启动的肺部肿瘤的转移的组织学。比例尺=50μm。f.单独肺部肿瘤中的多种hdr产生的致癌性kras等位基因。显示了具有每个等位基因的肿瘤的数量。未在任何肺部肿瘤中鉴定的等位基因未示出。图25.通过aav/cas9介导的hdr将突变型kras变体引入体细胞胰腺和肌肉细胞驱动了浸润性癌症的形成。a.将aav-krashdr/sgkras/cre逆行胰管注射至pt;h11lsl-cas9小鼠以诱导胰腺癌的示意图。b.通过将aav-krashdr/sgkras/cre逆行胰管注射至pt;h11lsl-cas9小鼠所启动的胰腺肿瘤的组织学。比例尺=75μm。c.患有原发性pdac的pt;h11lsl-cas9小鼠中的淋巴结(上图)和隔膜(下图)中的转移的组织学。比例尺=50μm。d.胰腺肿瘤块中的hdr产生的致癌性kras等位基因。显示了具有每个等位基因的肿瘤的数量。未在任何胰腺肿瘤块中鉴定的等位基因未示出。e.将aav-krashdr/sgkras/cre肌肉内注射至pt;h11lsl-cas9小鼠的腓肠肌以诱导肉瘤的示意图。f,g.通过将aav-krashdr/sgkras/cre肌肉内注射至pt;h11lsl-cas9小鼠的腓肠肌所启动的定型肉瘤(f)和浸润性肉瘤(g)的组织学。比例尺=75μm。h.肉瘤中的hdr产生的致癌性kras等位基因。显示了具有每个等位基因的肿瘤的数量。未在任何肉瘤中鉴定的等位基因未示出。这些数据记录了多个组织的细胞谱系的克隆标记。图26.使用单独的具有条形码的肿瘤的aav/cas9介导的体细胞hdr和高通量测序进行的kras突变型致癌性的多重、定量分析。a.通过肿瘤条形码的高通量测序来定量测量来自成批肺部样品的单独的肿瘤大小和数量的流程。b.归一化为初始呈现(aav质粒文库中的突变型呈现/aav质粒文库中的wt呈现)和相对于wt(突变型肿瘤数量/wt肿瘤数量)的具有每个突变型kras等位基因的肺部肿瘤的数量。与wt相比存在于显著更多的肿瘤中的变体(p<0.01)以蓝色表示;深蓝色表示与g12d相比无显著差异(p>0.05),浅蓝色表示与g12d相比具有该变体的肿瘤显著更少(p<0.01)。c.在不同基因型中具有每种kras变体的肺部肿瘤的数量的双侧多项式卡方检验的p值。显著性p值(p<0.05)为粗体。d,e.对于在所有lt;h11lsl-cas9(d)或pt;h11lsl-cas9(e)小鼠中在b中被鉴定为致癌性的kras变体的肺部肿瘤大小分布。每个点表示具有唯一的kras变体-条形码对的一个肿瘤。每个点的大小与它表示的肿瘤大小成比例,该大小通过将肿瘤读段计数归一化为归一化对照读段计数来估算。f.通过胰腺肿瘤块的肿瘤条形码测序来鉴定的多种hdr产生的kras等位基因。显示了具有每个等位基因的具有唯一条形码的肿瘤的数量。未在任何胰腺肿瘤块中鉴定的等位基因未示出。g.原发性胰腺肿瘤块的高通量测序和来自单只aav-krashdr/sgkras/cre-处理的pt;h11lsl-cas9小鼠的转移发现了多种突变型kras等位基因谱,并且能够建立原发性肿瘤及其转移后代之间的克隆关系。每个点表示具有指定的kras变体和指定样品中的唯一条形码的一个肿瘤。用彩色直线连接的点具有相同的条形码,表明它们是克隆相关的。每个点的大小根据它表示的肿瘤大小来缩放(点的直径=相对大小1/4)。由于胰腺肿瘤的大小未归一化为对照,因此只能在相同的样品内比较肿瘤大小。因此,每个样品中的最大肿瘤被设定为相同的标准大小。图27.针对kras的多重突变的aav文库的设计、产生和验证。a.靶向kras外显子2的三种sgrna的序列。通过在用编码每个sgrna的慢病毒载体转导后48小时对来自表达cas9的mef的dna的测序来测定每个sgrna的切割效率。全部三种sgrna均诱导靶基因座处的插入缺失形成。因此,靶向与kras密码子12和13最近的序列的sgrna(sgkras#3)被用于所有后续实验,以增加hdr的可能性。b.dsdna片段的合成文库,该dsdna片段含有野生型(wt)kras序列加上在密码子12和13处的12个非同义单核苷酸kras突变体中的每个、pam和sgrna同源区内的沉默突变(pam*)、以及用于单独肿瘤的条形码编制的下游密码子的摇摆位置内的8个核苷酸的随机条形码。每个kras等位基因可以与约2.4x104个唯一条形码相关。片段还含有用于克隆的限制性位点。c.c.aav载体文库通过以下方式产生:将合成区整体连接至亲本aav载体,以产生具有wtkras和kras密码子12和13中的全部12个单核苷酸非同义突变的具有条形码的库。d.krashdr模板内的kras外显子2的位置。显示了同源臂的长度。e.测试hdr偏差的实验的示意图。用aav-krashdr/sgkras/cre转导表达cas9的细胞系,然后对其进行测序以定量hdr事件。f.特异性扩增经由hdr引入基因组的krashdr等位基因的pcr策略的示意图。正向引物1(f1)结合至含有3个pam*突变的序列,而反向引物1(r1)结合krashdr模板的同源臂中存在的序列外部的内源性kras基因座。f2结合至通过f1添加的illumina衔接子,r2结合至接近外显子2的区,r3结合至在相同的反应中通过r2添加的illumina衔接子。g.在用aav-krashdr/sgkras/cre载体文库转导的培养物中通过表达cas9的细胞中的hdr产生的内源性kras基因座内的每个kras等位基因的呈现。h.在用于产生病毒文库的aav-krashdr/sgkras/cre质粒文库中针对每个kras突变型等位基因的初始频率绘制的每个krashdr等位基因的hdr事件的频率。在hdr后初始质粒文库和突变型kras等位基因的呈现之间的高度相关性表明很少有至几乎没有hdr偏差。图28.用于成体肺上皮细胞转导的最佳aav血清型的鉴定。a.筛选用于成体肺上皮细胞转导的11种aav血清型的实验概述。编码gfp的aav载体用不同的aav衣壳血清型包封,并且气管内施用于野生型接受者小鼠。在处理后5天,解离肺部并通过流式细胞术来测定gfp阳性上皮细胞的百分比。b.可以以不同的浓度产生不同的aav血清型。我们的目标是鉴定能够将dna模板递送至肺上皮细胞的aav血清型,这在很大程度上取决于可实现的病毒滴度和每个病毒粒子的转导效率二者。因此,在感染(转导)之前我们未对aav血清型的滴度进行归一化,而是在施用60μl未稀释的纯化病毒之后测定感染(转导)百分比。c.为了评估由不同的aav血清型转导的肺上皮细胞的百分比,我们将感染(转导)小鼠的肺解离成单细胞悬浮液,并针对gfp以及针对造血细胞(cd45、ter119和f4/80)、内皮细胞(cd31)和上皮细胞(epcam)的标志物进行流式细胞术。图显示了fsc/ssc设门的、活的(dapi阴性)肺上皮(cd45/ter119/f4-80/cd31阴性、epcam阳性)细胞。每个样品中gfp阳性上皮细胞的百分比在门上方显示。aav8、aav9和aavdj比所有其他血清型(包括不能产生有效hdr的aav6,载于platt等人,cell,2014)显著更好,与这些血清型的高最大滴度一致。我们根据该数据和已记录的aav8能够有效体内转导很多其他小鼠细胞类型的能力来选择使用aav8。图29.肺上皮细胞中aav/cas9介导的体内hdr启动可以进展以获得转移能力的原发性肿瘤。a.通过aav-krashdr/sgkras/cre的气管内施用来将点突变引入lkb1flox/flox;r26lsl-tomato;h11lsl-cas9(lt;h11lsl-cas9)、p53flox/flox;r26lsl-tomato;h11lsl-cas9(pt;h11lsl-cas9)和r26lsl-tomato;h11lsl-cas9(t;h11lsl-cas9)小鼠中的内源性kras基因座和条形码肺上皮细胞的实验的示意图。b.对应于图2a中的荧光图像的光学图像。更高放大率的组织学图像记录了p53缺陷型肿瘤中的腺癌组织学和更大的核非典型性。上图比例尺=5mm。下图比例尺=50μm。c.lt;h11lsl-cas9、pt;h11lsl-cas9和t;h11lsl-cas9小鼠中aav-krashdr/sgkras/cre诱导的肺部肿瘤的其他实例。比例尺=5mm。请注意,由于高转导效率,大多数肺部细胞表达tomato,但是由于每个肿瘤中的细胞数量和密度大,肿瘤更亮。d.具有用aav-krashdr/sgkras/cre启动的肿瘤的每种基因型小鼠中的肺总重量。每个点代表一只小鼠。e.在荧光解剖镜下鉴定的用1:10稀释的aav-krashdr/sgkras/cre感染(转导)的每种基因型小鼠中的表面肺部肿瘤的数量。每个点代表一只小鼠。f.在具有aav-krashdr/sgkras/cre启动的肺部肿瘤的pt;h11lsl-cas9小鼠中形成的淋巴微转移的组织学。比例尺=50μm。g.在胸膜腔(dtc>10)和淋巴结转移中具有弥散性肿瘤细胞的每种基因型小鼠的数量。该数量表示具有dtc或转移的小鼠的数量/所分析的小鼠的总数量。图30.无核酸酶的aav介导的hdr的发生率不足以启动大量肺部肿瘤。a.含有2.5kbkrashdr模板的对照aav载体文库的示意图,该模板具有12个单核苷酸、非同义突变和条形码,但是不具有靶向kras的sgrna。b.aav-krashdr/cre质粒库中的每个kras密码子12和13等位基因的呈现。百分比是一式三份测序的平均值。c.aav载体文库的滴度(vg=载体基因组)。重要的是,对照aav-krashdr/cre病毒制备物的滴度高于aav-krashdr/sgkras/cre。d.在施用60μl未稀释或1:10稀释的aav-krashdr/cre库后发展肿瘤的lt、pt和t小鼠的数量定量。图31.单独肿瘤的分析鉴定了致癌性kras等位基因并发现了非hdrkras等位基因中的插入缺失。a.具有pam*突变、g12d突变和条形码的krashdr等位基因的示例测序迹线。b.通过桑格(sanger)测序在单独的肺部肿瘤中检测到的四个代表性致癌性kras等位基因的序列。所分析的每种原发性肿瘤具有唯一的变体-条形码对,如预期的那样每个变体具有给定的约2.4x104个可能的条形码。显示了该基因座处的aav-krashdr模板序列和野生型kras序列中的改变的碱基以用于参考。c.hdr事件通常发生在两个工程改造的限制性位点的外部。然而,一些肿瘤具有与外显子2和一个限制性位点之间的重组一致的kras等位基因,这表明重组非常接近cas9/sgkras诱导的双链dna断裂。d.未经历完全hdr的单独肿瘤中的致癌性kras等位基因的示意图。完全和不完全hdr事件二者均存在于每种小鼠基因型中(完全hdr存在于lt;h11lsl-cas9小鼠的14/30的肿瘤和pt;h11lsl-cas9小鼠的3/7的肿瘤中)。不完全hdr事件包括可能通过外显子2上游的aav-krashdr模板的5’末端的同源重组以及aav-krashdr模板的3’末端至紧邻cas9/sgkras诱导的双链dna断裂下游的外显子2区的连接来整合进kras基因座的等位基因。该不完全hdr导致kras外显子2的内含子序列下游中的插入或缺失。插入和缺失的长度是可变的(通过桑格测序或凝胶电泳来估算的大小),并且有时包含野生型外显子2的部分或全部,或在极少数情况下包含aav-krashdr/sgkras/cre载体的区段。预期这些部分hdr事件均不会改变从突变体外显子2至外显子3的剪接,这与对于肿瘤形成的致癌性kras等位基因的表达要求一致。e,f.来自经处理的pt;h11lsl-cas9和lt;h11lsl-cas9小鼠的大型单独肿瘤中的致癌性kras等位基因几乎总是伴随着通过外显子2中的cas9介导的插入缺失形成产生的另一个kras等位基因的失活。桑格测序鉴定出邻近47/48(98%)的单独肿瘤中的pam序列的插入缺失。显示了示例插入缺失(e)和所有插入缺失的汇总(f)。nd表明不能检测到野生型等位基因,这与杂合性丧失、非常大的插入缺失或涵盖引物结合位点之一的大缺失中的任一者一致。图32.在胰腺细胞中hdr介导的致癌性突变引入内源性kras基因座导致形成胰管腺癌。a.将aav-krashdr/sgkras/cre逆行胰管注射至pt;h11lsl-cas9小鼠以诱导胰腺癌的示意图。b.在用aav-krashdr/sgkras/cre转导的pt;h11lsl-cas9小鼠中产生的胰腺肿瘤的代表性光学和荧光图像。比例尺=5mm。c.包括癌前panin病灶(左上图)、分化良好的肿瘤区域(右上图)和分化较差的pdac(左下图)的胰腺肿瘤进展的不同阶段的组织学图像。右下图显示了pdac内的富含胶原的间质环境(用三色染色)的发育。比例尺=75μm。d.代表性facs图,它显示了具有aav-krashdr/sgkras/cre启动的pdac的pt;h11lsl-cas9小鼠胸膜腔内的tomato阳性弥散性肿瘤细胞(dtc)。图显示了fsc/ssc设门的活癌细胞(dapi/cd45/cd31/f4-80/ter119阴性)。e.hdr诱导的pdac可以进展以获得转移能力,在淋巴结中和隔膜上接种转移。显示了光学和荧光解剖镜图像。比例尺=3mm。f.用指定的aav载体文库感染(转导)后3至13个月的pdac发生率、腹膜腔中的dtc以及指定基因型小鼠中的转移(以所分析的小鼠总数中具有癌症、dtc或转移的小鼠的数量显示)。图33.在骨骼肌中hdr介导的致癌性kras的诱导诱导肉瘤。a.将aav-krashdr/sgkras/cre肌肉内注射至pt;h11lsl-cas9小鼠的腓肠肌以诱导肉瘤的示意图。b.在用aav-krashdr/sgkras/cre注射后小鼠腓肠肌的代表性全组织光学标本包埋(上图)和荧光解剖镜(下图)图像。右边腓肠肌具有肉瘤,而左边则没有,尽管广泛的tomato阳性组织证实了有效转导(数据未示出)。比例尺=5mm。c.组织学h&e切片的图像证实了具有定型组织学的肉瘤的存在以及向周围肌肉的浸润。比例尺=75μm。d.在肌肉内注射aav-krashdr/sgkras/cre后3至7个月的pt;h11lsl-cas9小鼠中肉瘤的发生率。发生率表示为已注射的小鼠的总数中产生肉瘤的小鼠的数量。7只经处理的小鼠中的一只未进行分析,但是在感染(转导)后六个月没有明显的肉瘤。e.肉瘤中krashdr基因座的测序显示出突变型kras等位基因和条形码。图34.用于成批肺部组织的测序的样品和制备物,该测序用于定量具有每个突变型kras等位基因的肺部肿瘤的大小和数量。a.用于具有条形码的krashdr等位基因的测序的气管内施用有aav-krashdr/sgkras/cre的小鼠的成批肺部组织样品。显示了样品名称、小鼠基因型和aav-krashdr/sgkras/cre的稀释度。显示了解剖的肿瘤的重量、肿瘤数量、数量以及用于每个样品的测序的所扩增的dna量和合并的pcr反应的数量。重复样品是技术重复。nd=无数据。b.使用来自已知细胞数的基准对照的读段使来自成批肺部样品的测序读段的归一化,以便能够估算每个肿瘤中的细胞数以及允许来自单独小鼠的数据组合的简化流程。图35.来自成批组织的肿瘤基因型、大小和数量的基于条形码测序的平行分析的再现性。a-d.具有指定的krashdr等位基因和可通过技术重复(即来自成批组织裂解物和pcr反应的独立dna提取物)中的高通量测序检测到的唯一条形码的单独的肿瘤的回归图。使用具有不同的多重标签的引物对a和b中的重复进行pcr扩增,但是这些重复在相同的测序泳道上运行。使用相同的引物对c和d中的重复进行pcr扩增,但是这些重复在不同的测序泳道上运行。对肿瘤负荷高于平均水平(a、c)和肿瘤负荷低于平均水平(b、d)的小鼠(如通过成批肺部重量所测量)进行分析以确认在可变肿瘤数量的样品中该流程的技术和计算再现性。图36.来自成批肺部组织的高通量条形码测序发现了多种数量和大小的肿瘤。a-c.在全部lt;h11lsl-cas9(n=6)(a)、pt;h11lsl-cas9(n=7)(b)或t;h11lsl-cas9(n=3)(c)小鼠中所有kras变体的肿瘤大小分布。每个点表示具有唯一的kras变体-条形码对的肿瘤。每个点的大小与它表示的肿瘤大小成比例,该大小通过将肿瘤读段计数归一化为归一化对照读段计数来估算。具有wtkrashdr等位基因的病灶被认为是具有致癌性krashdr等位基因的肿瘤中的便车客(hitchhiker)(参见方法)。d,e.在每种基因型中具有每种kras变体的肿瘤的原始(d)和归一化(e)数量表(包括具有通过单独肿瘤解剖和分析所鉴定的每种变体的肿瘤)。在e中,具有每种kras变体的肿瘤的数量被归一化为每种变体在aav质粒文库中的初始呈现以及在相同基因型内具有wt等位基因的病灶的数量。请注意,为了便于比较,e中的热图的色彩强度量表是每种基因型唯一的。图37.胰腺肿瘤块和转移的高通量测序鉴定出致癌性kras突变体。a.来自通过逆行胰管注射施用aav-krashdr/sgkras/cre以对具有条形码的krashdr等位基因进行illumina测序的小鼠的成批胰腺组织和转移样品。显示了样品名称、小鼠基因型、病毒稀释度和组织。在从这些样品facs分离fsc/ssc设门的活癌细胞(dapi/cd45/cd31/f4-80/ter119阴性)后通过测序来分析原发性肿瘤块以及转移的独特区域中存在的krashdr等位基因。b.鉴定pt;h11lsl-cas9小鼠的胰腺内的aav-krashdr/sgkras/cre启动的肿瘤块中的krashdr等位基因的分析流程。c.单只aav-krashdr/sgkras/cre处理的pt;h11lsl-cas9小鼠中的大胰腺肿瘤块的多区域测序发现了多种突变型kras等位基因谱,并且将原发性肿瘤与它们的转移后代相关联。每个点表示具有指定的kras变体和指定样品唯一的条形码(标记为1-4)的肿瘤。共有相同的kras变体-条形码对的不同原发性肿瘤样品(标记为1-3)中的点被连接在一起,因此这些点可能是多个样品中存在的相同的原发性肿瘤的区域。彩色线将具有相同的kras变体-条形码对的原发性肿瘤和淋巴结转移连接在一起,它表示克隆关系。每个点的大小根据它表示的肿瘤大小来缩放(点的直径=相对大小1/2)。由于胰腺肿瘤的大小未归一化为对照,因此只能在相同的样品内将肿瘤大小与其他肿瘤进行比较。因此,每个样品内最大的肿瘤已缩放至相同的标准尺寸。g=胆囊,sto=胃,duo=十二指肠,pan=胰腺,sp=脾,in=肠系膜淋巴结。图38.kras突变体的体内致癌性和生物化学行为之间的关系。a-c.用aav-krashdr/sgkras/cre转导的小鼠中的肺部肿瘤的相对数量(参见图4b)随hunter等人,2015报道的指定生物化学性质的变化。相对肺部肿瘤数量被归一化为aav-krashdr/sgkras/cre质粒库中每种kras变体的初始呈现。垂直条表示针对归一化的相对肺部肿瘤数量的95%置信区间。水平表表示三个重复实验的平均值的标准误差,如hunter等人,2015所述。p120gap被用于测定gap刺激的gtp水解速率(hunter等人,2015)。d-f.用aav-krashdr/sgkras/cre转导的小鼠中的胰腺肿瘤的数量(参见图4f)随hunter等人,2015报道的指定生物化学性质的变化。垂直条表示针对胰腺肿瘤数量的95%置信区间。水平表表示三个重复实验的平均值的标准误差,如hunter等人,2015所述。p120gap被用于测定gap刺激的gtp水解速率(hunter等人,2015)。图39.对组合遗传改变的研究:在krasg12d驱动的肺部肿瘤中p53缺陷改变了体内肿瘤抑制的生长作用。a.研究体内组合肿瘤抑制失活的tuba-seq方法。在三种不同的基因工程小鼠背景:kraslsl-g12d/+;rosa26lsl-tdtomato;h11lsl-cas9(kt;cas9)、kt;p53flox/flox;cas9(kpt;cas9)和kt;lkb1flox/flox;cas9(klt;cas9)中,用lenti-sgts-pool/cre(含有四个sgrna载体和十一个靶向已知和候选肿瘤抑制基因的载体)来启动肿瘤。每个sgrna载体含有唯一的sgid和随机条形码,该载体用于经由深度测序来定量单独的肿瘤大小。b.在肿瘤启动后15周kt;cas9小鼠中的相对肿瘤大小分析。指定百分位处的相对肿瘤大小是来自10只小鼠的合并数据,这些数据已归一化为sginert肿瘤的平均大小。整个该研究中的误差条表示通过自举采样确定的95%置信区间。与sginert显著不同的百分位以彩色表示。c.假设肿瘤大小呈对数正态分布,平均肿瘤大小的估算值鉴定出使kt;cas9小鼠中的生长显著增加的sgrna。显示了邦费罗尼校正的自举p值。p值<0.05的sgrna以粗体表示。d,e.与b、c相同,但来自12只kpt;cas9小鼠的合并数据除外。f.靶基因座处的插入缺失相对于靶向基因组的无活性sgrnasneo1-3的中位数的丰度。根据a进行着色。g.来自tcga和genie数据集(n=1792)的人肺部腺癌中的tp53和rb1中的功能性突变。rb1和tp53改变共同发生。图40.对组合遗传改变的研究:在lkb1缺陷型肿瘤中肿瘤抑制物失活的减弱作用进一步突出显示了崎岖的适合度景观。a.相对于相同百分位处的含有sginert的肿瘤的平均大小而言,对于每个sgrna的指定百分位处的肿瘤大小。显示了用lenti-sgts-pool/cre进行肿瘤启动后15周来自13只kt;lkb1flox/flox;cas9(klt;cas9)小鼠的合并数据。与sginert显著不同的百分位以彩色表示。b.假设肿瘤大小呈对数正态分布,平均肿瘤大小的估算值鉴定出使klt;cas9小鼠中的生长显著增加的sgrna。显示了邦费罗尼校正的自举p值。p值<0.05的sgrna以粗体表示。c.来自tcga和genie数据集(n=1792)的人肺部腺癌中的lkb1(stk11)和setd2突变的相互排斥性。d.具有lenti-sgsetd2/cre启动的肿瘤的kpt;cas9小鼠(n=7)与具有lenti-sgneo2/cre启动的肿瘤的kpt;cas9小鼠(n=3)中的肿瘤大小。lenti-sgsetd2/cre启动的肿瘤具有比lenti-sgneo2/cre启动的肿瘤大2.4倍的ln平均值以及大4.6倍的第95个百分位的肿瘤大小。e.具有lenti-sgsetd2/cre启动的肿瘤的klt;cas9小鼠(n=7)与具有lenti-sgneo2/cre启动的肿瘤的klt;cas9小鼠(n=5)中的肿瘤大小。相对ln平均值和相对第95个百分位数为2.2和2.8,它们二者均显著小于图2d中的数值(分别地p<0.04和p<0.0001)。f.在遗传背景中肿瘤抑制物的适合度作用的皮尔逊(pearson)相关性(通过ln平均值来确定)。sgp53和sglkb1生长速率不包括在kpt;cas9和klt;cas9小鼠中。*p<0.05,****p<0.0001。g.在致癌性kras驱动的肺部肿瘤以及具有同时发生的p53或lkb1缺陷的背景下每个肿瘤抑制基因的差异作用。显著偏离sginert肿瘤的第95个百分位以蓝色显示。h.将候选肿瘤抑制物鉴定为驱动子(如g中所定义)的可能性与所研究的遗传背景的数量。对所有遗传背景取平均值。图41.用于本研究中的推定肿瘤抑制物改变和人肺部腺癌中的这些基因组改变的频率分析的肺癌的基因工程小鼠模型的当前状态。a.来自公开研究的数据汇总,其中此处研究的推定肿瘤抑制基因在具有或不具有p53或lkb1失活的致癌性kras驱动的肺癌模型的背景下是失活的。b.对于所有肿瘤(所有)以及对于tp53(tp53mut)或lkb1(lkb1mut)中具有潜在失活改变的肿瘤,每个肿瘤抑制基因中具有潜在失活改变(移码突变或非同义突变,或者基因组丧失)的肿瘤的百分比。显示了具有每种类型改变的肿瘤的百分比。显示了两项临床癌症基因组学研究的数据:癌症基因组图集(cancergenomeatlas,tcga,2014)和基因组学证据瘤形成信息交换(genomicsevidenceneoplasiainformationexchange,genie,2017)数据库。图42.体内定量肿瘤大小分布的多重慢病毒载体、肿瘤启动和tuba-seq流程的描述。a.lenti-sgts-pool/cre含有四个具有无活性sgrna的载体和十一个具有靶向肿瘤抑制基因的sgrna的载体。每个sgrna载体含有唯一的sgid和随机条形码。nt=非靶向。b.lenti-sgts-pool/cre中的载体的sgid-条形码区的示意图。lenti-sgts-pool/cre含有具有十五个不同的8个核苷酸的唯一标识符(sgid)的载体,该唯一标识符将给定的sgid-条形码读段连接至特定的sgrna。这些载体还含有15个核苷酸的随机条形码元件(例如,唯一分析标识符,umi)。该双条形码系统允许鉴定单独的肿瘤,以及启动每个肿瘤的载体中的sgrna。c.用具有条形码的lenti-sgts-pool/cre库转导肺上皮细胞在基因工程小鼠模型中启动了肺部肿瘤,该基因工程小鼠模型具有(1)cre调控的致癌性krasg12d(kraslsl-g12d/+)等位基因、(2)cre报告等位基因(rosa26lsl-tomato)、(3)cre调控的cas9等位基因(h11lsl-cas9)以及(4)p53或lkb1的纯合的floxed等位基因。慢病毒载体稳定整合进所转导的细胞的基因组中。在kt;cas9、kpt;cas9和klt;cas9小鼠中启动肿瘤,以产生31种不同基因型的肺部肿瘤。在肿瘤生长15周后分析小鼠。在添加具有条形码的“基准”细胞系后,从整个肺部提取基因组dna,对sgid-条形码区进行pcr扩增、深度测序和分析,以便使用tuba-seq流程来确定每种具有唯一条形码的肿瘤的相对扩增。图43.krasg12d驱动的肺部腺癌中的体内肿瘤抑制。a.kt;cas9小鼠相对于kt小鼠的sgid呈现中的倍数变化(δsgid呈现),所述kt小鼠缺乏cas9,因此相对于sginert不应扩增。若干sgrna(sgid)在呈现中增加,这反映了随着被靶向的肿瘤抑制基因的失活肿瘤生长增加。显示了平均值和95%置信区间。b,c.与成批sgrna呈现(δsgid呈现)相比,通过分析具有单独条形码的肿瘤可以改善检测肿瘤抑制效果的能力。(b)具有每个sgrna的第95个百分位肿瘤的相对大小的分析鉴定出与成批δsgid呈现稍微相似的相对肿瘤大小估算值,这表现出更广泛的置信区间。(c)相对肿瘤大小的对数正态平均值(ln平均值)度量的p值与p值δsgid呈现。由于首先测量单独的肿瘤大小然后将其适当归一化以消除外源性来源的噪声,因此第95个百分位和ln平均值两个指标均以更大的置信度和精度鉴定出功能性肿瘤抑制物。p53丧失是例外,因为它的生长作用较差,如对数正态分布所描述。所有p值都是双侧的并且经由针对所研究的肿瘤抑制物的数量的2×106自举置换检验和邦费罗尼校正来获得。d-f.与a-c中相同,但kpt;cas9小鼠中的生长作用除外。倍数变化相对于kt小鼠而言,而第95个百分位和ln平均大小估算值相对于kpt;cas9内部sginert对照而言。g-i.与a-c中相同,但klt;cas9小鼠中的生长作用除外。没有tuba-seq,就不能鉴定出肿瘤抑制物。图44.肺部腺癌中的rb和p53肿瘤抑制物协同性通过tuba-seq来鉴定,使用cre/lox调控的等位基因在小鼠模型中确认,并且得到rb1和tp53突变共存在于人肺部腺癌中的支持。a.sgsetd2、sglkb1和sgrb1肿瘤的相对ln平均大小。rb1失活引起的肿瘤大小的增加小于p53完整的kt;cas9背景下的setd2或lkb1失活。相反,rb1失活引起的肿瘤大小增加程度类似于p53缺失的kpt;cas9背景下的setd2或lkb1失活。p值检验了与sgrb1相似的ln平均值的零假设。p<0.05以粗体表示。b.具有用adeno-cmv/cre启动的肿瘤的kp和kp;rb1flox/flox小鼠的代表性肺叶的h&e染色。在肿瘤启动后12周分析小鼠。比例尺=500μm。c.显示了kp和kp;rb1flox/flox小鼠的肺部的代表性离体μct图像。肺叶的轮廓以白色虚线勾勒。d.k;rb1wt/wt、k;rb1flox/flox、kp;rb1wt/wt和kp;rb1flox/flox小鼠中的肿瘤面积百分比的定量。组织学定量证实,在p53缺陷型肿瘤中rb1缺失更显著地使肿瘤负荷增加。*p值<0.05,n.s.=无显著性。显示了ad-cre的滴度。e,f.rb1和tp53突变共存在于两个人肺部腺癌基因组学数据集中:(e)tcga2014数据集和(f)genie联盟2017。使用针对体细胞改变的discover统计学独立性检验来计算p值。图45.靶基因组基因座的深度测序证实在所有靶基因座处产生了插入缺失,并且显示出最强肿瘤抑制基因中具有插入缺失的癌细胞的选择性扩增。a.sgrna靶向的每个区域中的插入缺失丰度,如通过来自四只kpt;cas9小鼠的被靶向的区域的总肺dna的深度测序所测定。插入缺失丰度被归一化为sgneo1、sgneo2和sgneo3的中位数丰度。误差条表示观察到的丰度范围,而点表示中位数。在所有被靶向的区域中观察到插入缺失。未示出sgp53,因为它的靶位点已通过cre介导的p53floxed等位基因的重组而缺失。b.(a)中所述的插入缺失丰度与通过tuba-seq测定的第95个百分位的肿瘤大小(如图1d中所述)。每个点表示单独的小鼠中的单个sgrna,并且每只小鼠以唯一的形状表示。插入缺失丰度与tuba-seq大小谱相关(正如预期的那样),然而插入缺失丰度不能测量单独的肿瘤大小并且表现出更大的统计噪声。如通过tuba-seq所测定,在整个该分析中的最大的单个肿瘤是在插入缺失分析中类似地显示为异常的sgcdkn2a肿瘤—进一步证实了通过tuba-seq进行的遗传事件的忠实分析。图46.小鼠模型和人肺部腺癌中setd2和lkb1之间的冗余度验证。a.来自具有lenti-sgsetd2#1/cre或lenti-sgneo2/cre启动的肿瘤的kpt和kpt;cas9小鼠的肺叶的荧光解剖镜图像(上图)和h&e染色切片(下图)。在肿瘤生长9周后分析小鼠。在荧光解剖镜图像中,肺叶轮廓以白色虚线勾勒。上图比例尺=5mm。下图比例尺=4mm。b.具有lenti-sgsetd2#1/cre或lenti-sgneo2/cre启动的肿瘤的kpt;cas9小鼠和具有lenti-sgsetd2#1/cre启动的肿瘤的kpt小鼠中的肿瘤面积百分比的定量。每个点表示小鼠,并且水平条是平均值。具有用相同的病毒启动的肿瘤的kpt;cas9和kpt小鼠之间的肿瘤面积增加,但是具有用lenti-sgsetd2#1/cre启动的肿瘤的kpt;cas9小鼠和具有用lenti-sgneo2/cre启动的肿瘤的那些小鼠之间无差异,这可能是由于较高的小鼠间差异。因为这些慢病毒载体是具有条形码的,所以我们对这些小鼠进行tuba-seq分析,以定量所诱导的肿瘤大小。相对于sgneo2而言,sgsetd2使kpt;cas9中的肿瘤大小增加。**p<0.01,n.s.为无显著性。c,d.与a,b相同,但是具有lenti-sgsetd2#1/cre或lenti-sgneo2/cre启动的肿瘤的klt;cas9小鼠除外。在肿瘤生长9周后分析小鼠。上图比例尺=5mm。下图比例尺=4mm。e,f.setd2和lkb1(hgnc名称stk11)共存在于两个人肺部腺癌基因组学数据集中:(e)tcga2014数据集14(n=229名患者)和(f)genie联盟(n=1563名患者)。使用discover统计学独立性检验来计算双侧p值。图47.tuba-seq适合度测量与人基因组模式的对应性。a.我们所研究的十九个成对相互作用的相对适合度测量和人共存在率。ln平均值率是所关注的背景内的相对ln平均值(sgts/sginert)除以全部三个背景的平均相对ln平均值的比率。背景率可以是三个背景的未加权平均值(原始),或存在于人肺部腺癌中的每个背景的比率的加权平均值(加权)。*or=人数据中的基因对的共存在率的“优势率”。使用discover检验来确定人共存在率的单侧p值(>0.5表示相互排斥)。使用史托佛(stouffer)法来产生组合p值(方法)。p<0.025和p>0.975以粗体表示。适合度测量和共存在率通常是对应的(对于加权ln平均值比率,斯皮尔曼(spearman)r=0.50,p值=0.03;对于未加权ln平均值比率,r=0.4)。b.通过组合优势率>1和冗余度<1来定义来自a.人遗传协同性的适合度测量和共存在率的图形汇总。针对tuba-seq数据的协同性表示ln平均值比率>1和冗余度<1。c.来自二十一种肿瘤类型的泛癌分析表明了统计学显著性遗传相互作用的数量。肿瘤类型缩写借用自tcga。肺部腺癌(luad)以黑色表示,并且预计含有大量与中位数相似的遗传相互作用,这表明此处研究的适合度景观的崎岖度通常可以代表癌症进化。图48.较大遗传调查的功效分析。通过假设肿瘤大小呈对数正态分布,检测较大遗传调查中的驱动子生长作用和非加性驱动子相互作用的tuba-seq的统计学功效可以被映射。未来的实验可以采用较大的小鼠队列和较大的靶向推定肿瘤抑制物的sgrna库。在所有假设实验中,具有无活性sgrna的库的lenti-sgts-pool/cre滴度和分数(用于归一化)与我们的原始实验保持一致。a.针对检测弱驱动子中的置信度的p值等高线(通过kt;cas9小鼠中的sgcdkn2a分布来参数化)。大于等高线的任何实验设置都会检测到置信度大于或等于等高线的p值的弱驱动子。b,c.与a中相同,但是分别地中等驱动子和强驱动子除外(通过kt;cas9小鼠中的sgrb1和sglkb1来参数化)。sgrna库大小可扩大为500个靶标(而不是库中的100个靶标),因为当研究具有这些作用强度的基因时,可以进行更大的筛选。d-f.与a-c中相同,但驱动子相互作用除外。驱动子相互作用(ln平均值比率)被定义为在统计学上不同于值为一的零假设的驱动子生长率的比率(背景#1中的sgts/sginert)/(背景#2中的sgts/sginert)。(d)通过rbm10—p53参数化的弱驱动子相互作用(7%的作用大小)。(e)通过rb1—p53参数化的中等驱动子相互作用(13%的作用大小)。(f)通过setd2—lkb1参数化的强驱动子相互作用(68%的作用大小)。图49.揭示肺癌疗法的kras基因型特异性的方法。联合crispr/cas9辅助的hdr和治疗性治疗与基于测序的肿瘤尺寸定量,以产生基因型-药物反应矩阵(5,上)。指示了药物基因组学分析流程的时间表(1-5,下)。加圆圈的数字对应于该实验的主要步骤。ptx=紫杉醇,carbo=卡铂,meki=mek抑制剂(曲美替尼)。图50.使用诱导型flp介导的慢病毒编码的sgrna的表达依次灭活一组肿瘤抑制基因的概述。(a)具有嵌入u6启动子中的tatafrt侧翼终止盒的pinsane载体。tatafrt位点内的tata框为粗体。flp活性移除了终止盒并启动sgrna表达。指示了通用染色质开放元件(ucoe)和sgid/bc区域。(b)insane-sgts-pool/cre含有靶向11个肿瘤抑制物和4个inertsgrna的sgrna。(c)实验组。指示了两个阴性对照队列和两个阳性对照队列。指示了他莫昔芬(tam)治疗的时间。图51.体内肿瘤抑制基因的组合双重sgrna靶向。(a)用于表达两个sgrna的我们的病毒载体的示意图。包括锚定sgrna的七个带条形码的载体库(sginert或靶向p53或lkb1的六个sgrna之一)和靶向11种肿瘤抑制物的sgrna库。(b)肿瘤抑制物配对的多重分析联合tuba-seq分析的示意图。图52.将癌基因标识条形码并入基因内含子中的策略。说明了kras的内含子2的情况,在小鼠体内生成多个不同的致癌激活突变的构建体连同标识突变的条形码编制。在构建体中包括靶向kras的内含子2的sgrna,以及跨越kras的激活突变热点和kras的内含子2两者的hdr盒。hdr盒携带激活突变(在其外显子部分中)、pam突变(以防止再次裂解修复的转录物)和条形码序列(在其内含子部分中)。条形码序列包括唯一地标识所引入的突变的区段,以及任选地标识产生肿瘤的单独核酸分子的唯一分子标识符序列。具体实施方式在描述本发明的方法和组合物之前,应当理解,本发明不限于所描述的特定方法或组合物,因此这些方法或组合物当然是可变的。还应当理解,本文所用的术语仅出于描述特定实施方案的目的,并不旨在具有限制意义,因为本发明的范围将仅受所附权利要求的限定。在提供值的范围时,应当理解,除非上下文另外明确指出,否则还明确公开了介于该范围上限与下限之间的每个居间值(至下限单位的十分之一)。在规定范围内的任何规定值或居间值与在该规定范围内的任何其他规定或居间值之间的每个较小范围都涵盖在本发明内。这些较小范围的上限和下限可以独立地包括在该范围内或排除在该范围之外,并且在任一个限值、两个限值都不或两个限值都包括在较小范围内的情况下,每个范围也涵盖在本发明内,以规定范围内任何特别排除的限值为依据。当规定范围包括一个或两个限值时,排除了那些所包括的限值中的任一个或两个的范围也包括在本发明中。除非另外定义,否则本文使用的所有技术和科学术语均具有与本发明所属领域中的普通技术人员通常所理解的相同含义。尽管与本文描述的那些类似或等同的任何方法和材料也可用于本发明的实践或试验,但现在描述的是一些潜在和优选的方法和材料。本文提及的所有出版物均通过引用并入本文,以公开和描述与所引用的出版物有关的方法和/或材料。应当理解,在存在矛盾的方面来说,本公开取代所并入的出版物的任何公开内容。正如对于本领域的技术人员而言在阅读本公开后将显而易见的,本文描述和展示的每个单独实施方案具有分立的组分和特征,这些组分和特征在不脱离本发明的范围或精神的前提下,可以容易地与任何其他几个实施方案的特征分离或组合。任何叙述的方法都可以按照叙述的事件的顺序或按照逻辑上可能的任何其他顺序进行。必须注意,如本文和所附权利要求中所用,除非上下文另外明确指出,否则单数形式“一个”、“一种”和“所述”包括复数指代物。因此,例如涉及“一个细胞”包括多个此类细胞(例如,一群此类细胞),并且涉及“所述蛋白”包括涉及一个或多个蛋白以及本领域的技术人员已知的它们的等同物(例如,多肽)等等。本文讨论的出版物仅仅是为了它们在本申请的提交日期之前的公开而提供的。本文的任何内容均不应解释为承认本发明无权凭借在先前发明而先于此类出版物。此外,提供的出版日期可能与实际出版日期不同,可能需要独立确认。方法和组合物如上文所概述,提供了用于测量同一个体中的多个克隆细胞群的群体大小的组合物和方法。作为一个实例,在一些情况下,主题方法是测量同一个体的多个克隆独立的肿瘤细胞群(例如,不同的肿瘤)的肿瘤大小(例如,肿瘤内的赘生性细胞的数量)的方法。在一些情况下,主题方法包括:(a)使个体的组织与可遗传且可彼此区分的多种细胞标志物接触,以在所接触的组织中产生可遗传标记的细胞的多种可区分的谱系。(b)在对于至少一部分可遗传标记的细胞经历至少一轮分裂而言已过去足够的时间之后,检测和测量所接触的组织中存在的多种细胞标志物中的至少两种的数量,从而产生一组测量值。以及(c)使用所述一组测量值作为输入来计算对于可遗传标记的细胞的至少两种所述可区分的谱系而言在所接触的组织中存在的可遗传标记的细胞的数量。与组织接触在一些实施方案中,主题方法包括使组织(例如,个体的组织)(例如,肌肉、肺、支气管、胰腺、乳腺、肝、胆管、胆囊、肾、脾、血液、肠、脑、骨、膀胱、前列腺、卵巢、眼、鼻、舌、口、咽、喉、甲状腺、脂肪、食管、胃、小肠、结肠、直肠、肾上腺、软组织、平滑肌、脉管系统、软骨、淋巴、前列腺、心脏、皮肤、视网膜以及繁殖和生殖系统(例如,睾丸、繁殖组织)等等)与可遗传且可彼此区分的多种细胞标志物接触的步骤,以在所接触的组织中产生可遗传标记的细胞的多种可区分的谱系。在一些情况下,组织是在动物体外生长的经工程改造的组织(例如,类器官、培养物中的细胞等)。在一些情况下,组织是活动物的一部分,所以组织可以被视为个体的组织,并且所述接触可以通过将细胞标志物施用(例如,经由注射)于个体来进行。可以使用任何方便的施用途径(例如,气管内施用、鼻内施用、逆行胰管施用、肌肉内施用、静脉内施用、腹膜内施用、囊内施用、关节内施用、局部施用、皮下施用、口服施用、瘤内施用等等)。在一些情况下,施用经由注射(例如,直接向靶组织注射文库(诸如病毒文库))来进行。在一些情况下,标志物向细胞的转移经由电穿孔(例如,核转染)、转染(例如,使用磷酸钙、阳离子聚合物、阳离子脂质等)、流体动力学递送、声孔效应、生物弹道颗粒递送或磁转染来进行。可以使用任何方便的递送载体(例如,病毒颗粒、病毒样颗粒、裸核酸、质粒、寡核苷酸、外泌体、脂质复合物、囊泡(gesicle)、聚合物体、多复合物、树状聚合物、纳米颗粒、生物弹道颗粒、核糖核蛋白质复合物、树状聚合物、细胞穿透肽等)。组织可以是来自任何所需的动物的任何组织类型。例如,在一些实施方案中,所接触的组织是无脊椎动物组织(例如,蜕皮动物、冠轮动物、多孔动物、刺胞动物、栉水母动物、节肢动物、环节动物、软体动物、扁形动物、轮形动物、节肢动物、昆虫或蠕虫组织)。在一些实施方案中,所接触的组织是脊椎动物组织(例如,禽、鱼、两栖动物、爬行动物或哺乳动物组织)。合适的组织还包括但不限于来自以下各项的组织:啮齿动物(例如,大鼠组织、小鼠组织)、有蹄类动物、农场动物、猪、马、奶牛、绵羊、非人灵长类动物和人。靶组织可以包括但不限于:肌肉、肺、支气管、胰腺、乳腺、肝、胆管、胆囊、肾、脾、血液、肠、脑、骨、膀胱、前列腺、卵巢、眼、鼻、舌、口、咽、喉、甲状腺、脂肪、食管、胃、小肠、结肠、直肠、肾上腺、软组织、平滑肌、脉管系统、软骨、淋巴、前列腺、心脏、皮肤、视网膜以及繁殖和生殖系统(例如,睾丸、繁殖组织)等等。在一些情况下,出于将细胞诱导为赘生性细胞的目的而接触组织,例如在一些情况下,出于启动形成多个独立的肿瘤的目的而接触组织。例如,在一些情况下,所引入的细胞标志物(和/或与细胞标志物连接的组分)导致赘生物转化(导致赘生性细胞形成),并且可以对多个不同的赘生物启动事件的结局进行彼此比较,因为每个事件是用可识别的可遗传细胞标志物唯一标记的。在一些此类情况下,细胞标志物启动相同的遗传变化,以使得所诱导的肿瘤由于相同类型的(或甚至相同的)遗传干扰而起始,但是可以追踪每个启动事件的结局,因为每个单独的细胞标志物可彼此区分。这种方法的目的可以是例如追踪相同的组织(和/或相同的动物)中的多个独立的细胞谱系,以产生对于所关注的给定基因型的群体大小(例如,肿瘤大小、每个肿瘤中的赘生性细胞数)分布谱。或者,在一些情况下,使用不同的遗传干扰(例如,细胞标志物可以导致两种或更多种不同的遗传干扰,与细胞标志物相连接的组分可以导致两种或更多种不同的遗传干扰),并且可以对相同的组织中(例如,在一些情况下,相同的动物中)的不同基因型的结局进行比较(例如,相同的组织中存在的具有不同的遗传基础的不同的肿瘤,例如肺、肌肉、肾等等中的多个不同的肿瘤)。在一些实施方案中,在与细胞标志物接触之前,组织已经含有赘生性细胞(例如,肿瘤)。在一些情况下,肿瘤与细胞标志物接触(例如,细胞标志物可以注射至肿瘤中,注射至血流中以接触一种或多种肿瘤,施用于另一个器官或组织以接触一种或多种肿瘤等)。作为一个实例,在一些情况下,细胞标志物被用作标记独立的赘生性细胞(诸如赘生物或肿瘤中的不同细胞)的方式,并且然后每个所标记的细胞可以被处理为单独的谱系–该谱系可以通过在一轮或多轮细胞分裂后对存在每个标志物的细胞的数量进行计数来追踪针对每个追踪的谱系而产生的细胞的数量。在一些情况下,方法包括对细胞标志物所引入的细胞进行遗传修饰。例如,在进行主题方法前组织可以已经具有一个或多个肿瘤,并且引入细胞标志物的目的是测试向肿瘤细胞引入另外的遗传修饰的效果(即,除已经存在于赘生性细胞中的变化之外的变化)。因此,每个可区分的细胞标志物可以与不同的遗传变化相关(例如,通过使编码指导rna(所述指导rna靶向具有唯一标识符(诸如dna条形码)的特定的遗传靶标)的核酸配对,以使得每个指导rna,从而每个遗传修饰与唯一标识符(诸如dna条形码)相关)。在这种情况下,所标记的谱系表示在遗传上彼此不同(例如,在特定遗传基因座处具有突变)的细胞组。或者,在一些情况下,每个肿瘤在遗传上是相同的,并且细胞标志物追踪在遗传上不一定彼此不同的谱系。这允许方法的执行者追踪相同的动物中的多个独立的细胞谱系,以及产生对于所关注的给定基因型的群体大小(例如,肿瘤大小、肿瘤中的赘生性细胞的数量)分布谱。细胞标志物多种细胞标志物(即,所引入的(异源性的、人工的)细胞标志物–其中所述标志物不是细胞中预先存在的那些标志物–例如,所引入的标志物并非仅仅是肿瘤中预先存在的克隆体细胞突变)是两种或更多种(例如,3种或更多种、5种或更多种、10种或更多种、或者15种或更多种、50种或更多种、100种或更多种、200种或更多种、500种或更多种、1000种或更多种、10000种或更多种、100000种或更多种、1000000种或更多种、1000000000种或更多种等)细胞标志物。同样,多个标记的细胞谱系是两个或更多个(例如,3个或更多个、5个或更多个、10个或更多个、或者15个或更多个、100个或更多个、1000个或更多个、10000个或更多个、100000个或更多个等)标记的细胞谱系。可以使用任何方便的可遗传细胞标志物(可彼此区分的),并且许多可遗传细胞标志物将是本领域的普通技术人员已知的。在一些情况下,细胞标志物(即,可遗传且可彼此区分的所引入的(异源性的、人工的)细胞标志物)是具有条形码的核酸。在一些情况下,具有条形码的核酸可以整合进靶细胞的基因组,或者在一些情况下,具有条形码的核酸可以维持在附加体中。具有条形码的核酸包括提供对于每个细胞谱系的唯一标识符的核苷酸序列,该唯一标识符将是能够检测和定量/测量的。在一些情况下,可遗传且可彼此区分的多种细胞标志物是具有条形码的核酸的文库,其中条形码的确切序列具有一些随机元件。例如,在一些情况下,条形码可以用一系列n来描述(例如,核酸序列中的位置,对于该位置每个核苷酸未确定,但是为规范或非规范核苷酸的所有可能的或确定的子集中的一者)。具有条形码的主题核酸可以包含任何方便的n的数量。在一些情况下,具有条形码的主题核酸(多个核酸/文库)包含5个或更多个(例如,6个或更多个、7个或更多个、8个或更多个、10个或更多个、12个或更多个或者15个或更多个)随机位置,例如5个或更多个(例如,6个或更多个、7个或更多个、8个或更多个、10个或更多个、12个或更多个或者15个或更多个)核苷酸未预先确定的位置。在一些情况下,具有条形码的核酸的文库(多个核酸)的公式包含长度为至少10个碱基对(bp)(例如,长度为至少12bp、15bp、17bp或20bp)的核苷酸的序列段,其中5个或更多个位置(例如,6个或更多个、7个或更多个、8个或更多个、10个或更多个、12个或更多个或者15个或更多个位置)未确定(即,在文库的成员中碱基同一性不同的位置)。在一些情况下,具有条形码的核酸的文库(多个核酸)的公式包含其中5至40个位置(例如,5至30、5至25、5至20、5至18、5至15、5至10、8至40、8至30、8至25、8至20、8至18、8至15、8至10、10至40、10至30、10至25、10至20、10至18、10至15、12至40、12至30、12至25、12至20、12至18或12至15个位置)未确定(即,在文库的成员中碱基同一性不同的位置)的核苷酸的序列段。在一些情况下,具有条形码的核酸的文库(多个核酸)的公式包含其中5至1000个位置(例如,5至800、5至600、5至500、5至250、5至150、5至100、5至50、5至30、5至25、5至20、5至18、5至15、5至10、8至1000、8至800、8至600、8至500、8至250、8至150、8至100、8至50、8至40、8至30、8至25、8至20、8至18、8至15、8至10、10至1000、10至800、10至600、10至500、10至250、10至150、10至100、10至50、10至40、10至30、10至25、10至20、10至18、10至15、12至1000、12至800、12至600、12至500、12至250、12至150、12至100、12至50、12至40、12至30、12至25、12至20、12至18或12至15个位置)未确定(即,在文库的成员中碱基同一性不同的位置)的核苷酸的序列段。具有条形码的核酸可以是线性(例如,病毒)或环状(例如,质粒)dna分子。具有条形码的核酸可以是单链或双链dna分子。非限制性实例包括质粒、合成核酸片段、合成的寡核苷酸、小环和病毒dna。具有条形码的核酸可以是rna分子、dna(dna分子)、rna/dna杂合体或核酸/蛋白质复合物。在一些情况下,细胞标志物可以包括可遗传且可彼此区分的多种生物标志物(例如,抗体、荧光蛋白、细胞表面蛋白),这些生物标志物是单独的或者当组合使用时与可彼此区分的以及可与多个其他生物标志物区分的多个相同或不同类型的其他生物标志物组合。在这种情况下,生物标志物可以以预定或随机的方式存在于单独的细胞和/或细胞谱系的内部或外部,并且可以使用本领域的普通技术人员通常已知的方法(例如,高通量/下一代dna测序、显微镜检查、流式细胞术、质谱分析等)来定量和/或测量。可以使用任何方便的方法来将细胞标志物递送至细胞。在一些情况下,细胞标志物(例如,具有条形码的核酸)经由病毒载体递送至组织。可以使用任何方便的病毒载体,并且实例包括但不限于:慢病毒载体、腺病毒载体、腺相关病毒(aav)载体、博卡病毒载体、泡沫病毒载体和逆转录病毒载体。在来自以下工作实例的一个实例中(参见图4a),多种细胞标志物经由慢病毒载体递送至靶组织。使用这样的慢病毒颗粒文库,其中每个病毒颗粒包含含有双组分条形码的一个具有条形码的核酸,其中第一组分是每个编码的指导rna唯一的,并且第二组分是每个分子唯一的,继而使得该核酸是待检测和定量/测量的每个细胞谱系唯一的。条形码的第二组分的序列的公式为nnnnnttnnnnnaannnnn。因此,在19个碱基对的序列段中,它们中的15个碱基对未确定(例如,随机化的)。文库中的每个具有条形码的核酸:(i)编码crispr/cas指导rna;(ii)包含第一条形码—与指导rna连接的唯一标识符8个核苷酸的条形码,以使得每个不同的指导rna序列连接至其自身的唯一的8个核苷酸的条形码;(iii)包含第二条形码—上述具有15个未确定的位置的随机的19个核苷酸的条形码[用于追踪细胞谱系];以及(iv)编码基因编辑蛋白(cre),该蛋白的表达引起靶组织中的cas9表达。因此,在这种情况下,多个具有条形码的核酸的多个不同的成员包含相同的第一条形码,其中每个第一条形码具有“对应的”指导rna。然而,第二条形码是文库中的每个成员唯一的,以使得将检测和定量/测量的每个细胞谱系具有唯一标识符。因此,虽然一些成员共有第一条形码序列,因为它们共有共同的指导rna,但是文库中的每个成员具有可用于追踪每个整合(即,每个谱系)的唯一的第二条形码。在一些情况下,可遗传和可区分的多种细胞标志物与可遗传且可彼此区分以及可与相关的另外多种细胞标志物中的细胞标志物区分的多种细胞标志物中的一种或多种(例如,1种或更多种、2种或更多种、3种或更多种、5种或更多种、7种或更多种、9种或更多种、11种或更多种、13种或更多种、15种或更多种或者20种或更多种)相关。例如,一个具有条形码的核酸可以包含四组分条形码,其中第一组分是候选疗法(例如,候选抗癌化合物)唯一的,第二组分是每个个体(例如,可以接受或可以不接受候选疗法的小鼠)唯一的,第三组分是所编码的指导rna唯一的,第四组分是每个分子唯一的,继而使得具有条形码的核酸将是待检测和定量/测量的每个细胞谱系唯一的。因此,在该实例中,每个细胞谱系中的细胞数可以被定量/测量,并且每个细胞谱系还可以通过其四组分核酸条形码直接连接至该细胞谱系中指导rna诱导的特定遗传干扰、该细胞谱系遇到的特定候选疗法、以及该细胞谱系所在的特定个体(例如,小鼠)。在一些情况下,条形码被掺入dna供体模板中,以用于同源定向修复(hdr)或例如将确定的核酸序列掺入基因组中所期望的位置的任何其他机制。例如,hdr修复模板可以用于将相同的编码变化(例如,相同的编码等位基因)或甚至所期望的变化的子集引入其接触的细胞的基因组中,但是每个整合事件可以独立地标记,因为hdr模板的文库已在特定的位置随机分配。在来自以下工作实例的一个实例中(参见例如图23a),多种细胞标志物(其中每个aav颗粒包含一个hdr模板的aav颗粒文库)通过aav颗粒递送至靶组织。每个aav中的hdr模板包含kras密码子12和13或野生型kras序列中的12个可能的非同义、单核苷酸点突变之一以及邻近的密码子的摇摆位置中的随机的8个核苷酸的条形码,从而唯一标记经历hdr的每个细胞。条形码为(n)gg(n)aa(r)tc(n)gc(n)ct(n)ac(n)at(h)(seqidno:1),因此为其中8个位置未确定的22个碱基对的序列段。在一些情况下,细胞标志物可以响应于外部干扰(例如,候选抗癌疗法)而接触组织。在这种情况下,外部干扰原(perturbagen)的施加可以随机地、以可调的概率或作为信号的组合匹配的结果而出现(例如,细胞的预定生理状态、特定基因、一组基因或多组基因的表达水平、一条或多条特定途径的活性水平和/或细胞或细胞谱系内部或外部的其他信号[例如,组织的身份、血液供应水平、整个个体的免疫状态、细胞的物理位置等])。例如,细胞标志物(例如,具有条形码的dna)可以在指导rna在特定类型的上皮细胞特异性的增强子的控制下表达以及cas9响应于施用于组织所存在的个体的化合物表达时接触组织。在一些情况下,细胞标志物可以体内接触单个活生物体中,或体外接触培养物或类器官培养物中的细胞群中的健康或患病细胞群或组织。在一些情况下,细胞标志物可以接触数量增加或减少或静态的赘生性细胞谱系。在一些情况下,细胞标志物可以响应于药物或其他生理学或环境干扰的施加、随机地以可调的概率、或经由在一定数量的细胞分裂后诱导细胞标志物接触组织的计数机制、准确地或随机地以可调的平均值和方差和其他矩、或作为信号的组合匹配的结果而接触组织。靶细胞的遗传修饰(改变)如上文所述,在一些实施方案中,方法包括对细胞标志物所引入的细胞进行遗传修饰。在一些此类情况下,所引入的细胞标志物是遗传修饰剂。例如,在一些情况下,细胞标志物是诱导遗传修饰(例如,基因组修饰)的具有条形码的核酸,并且在一些此类情况下是诱导赘生性细胞形成的具有条形码的核酸。例如,来自具有条形码的核酸的rna(例如,指导rna)和/或蛋白(例如,cre、crispr/casrna指导的蛋白等)的表达可以产生一个或多个基因组改变,并且在一些情况下,基因组改变导致靶细胞转化至赘生性细胞中(例如,在一些情况下,它可以导致肿瘤形成)。然而,细胞标志物(例如,具有条形码的核酸)是否引入基因组修饰可以与该细胞标志物是否可以诱导赘生性细胞形成无关。例如,在一些情况下,具有条形码的核酸可以编码癌基因(当表达为蛋白时可以导致赘生性细胞形成的基因)。在一些此类情况下,具有条形码的核酸不诱导靶细胞中的基因组变化,但是由于癌基因的表达而诱导赘生性细胞形成。在一些情况下,癌基因编码当蛋白过表达时可以导致细胞变为赘生性的野生型蛋白。在一些情况下,癌基因编码当蛋白表达时可以导致细胞变为赘生性的突变的蛋白(例如,kras的突变形式)。在一些情况下,细胞标志物(例如,具有条形码的核酸)将基因组修饰引入靶细胞中,但是该修饰可以与在细胞标志物和相关的基因组修饰的引入之前、期间或之后的一段时间出现的一个或多个另外的基因组修饰组合仅诱导赘生物形成(例如,肿瘤/癌症形成)。在另一个方面,在一些情况下,细胞标志物(例如,具有条形码的核酸)将基因组修饰引入靶细胞中,但是该修饰不诱导赘生物形成(例如,肿瘤/癌症形成)。例如,在一些情况下,具有条形码的核酸以无活性方式整合进靶细胞的基因组中。在一些情况下,具有条形码的核酸编码蛋白(例如,野生型或突变型蛋白),其中该蛋白不一定涉及癌症,例如一种或多种蛋白可以参与所关注的任何生物学过程,并且它的表达可以对细胞增殖和/或赘生性细胞形成产生作用(例如,可以不是癌基因或肿瘤抑制物)。在一些此类情况下,核酸整合进靶细胞的基因组,并且在其他情况下,核酸不整合进基因组(例如,可以维持在附加体中)。在一些情况下,具有条形码的核酸编码野生型或突变型蛋白,例如编码对肿瘤有害(例如,除生长/增殖控制之外的某种方式)的蛋白的cdna。在一些实施方案中,主题细胞标志物(例如,具有条形码的核酸)将基因组修饰引入靶细胞中,并且还诱导赘生性细胞形成(例如,肿瘤/癌症形成)。例如,在一些情况下,带条形码的核酸可在靶基因座处引起编辑以修饰肿瘤抑制物,改变癌基因的表达,编辑基因(例如,kras)成为赘生性诱导等位基因等。在一些实施方案中通过涉及癌基因或肿瘤抑制基因的基因组修饰,细胞标志物诱导赘生物形成。在一些实施方案中,涉及癌基因的基因组修饰是从并入的携带具有激活突变的癌基因的转基因上切除终止密码子,其中通过在基因的开放阅读框的起点并入[重组酶位点]-[终止密码子]-[重组酶位点]序列(例如loxp-终止密码子-loxp)而阻断转基因的表达,使得使得重组酶(例如,在loxp的情况下为cre)的表达导致去除终止密码子并激活癌基因的转录。重组酶位点可以是适于构建转基因动物的任何重组酶位点,诸如翻转酶(flp)、cre、dre、φc31整合酶、kd酵母重组酶、r酵母重组酶、b2酵母重组酶或b3酵母重组酶的重组酶位点。在一些实施方案中,涉及癌基因的基因组修饰是内源性癌基因中的激活突变(通过crispr裂解基因,然后通过同源定向修复hdr而引入的)。在一些实施方案中,激活突变伴有在激活癌基因突变上游或下游的前间区序列邻近基序(pam)突变和密码子(至少3、4、5、6、7、8、9或10个密码子)的摆动碱基的突变。在一些实施方案中,激活突变伴有在癌基因的内含子内部的前间区序列邻近基序(pam)突变和至少3、6、9、12、15、18或20个核苷酸的突变,使得癌基因的剪接不被破坏。在图52中展示了将此类条形码并入到kras的内含子中的说明。无论涉及癌基因的基因组修饰是转基因还是内源性癌基因的突变,癌基因都可以是证明在细胞模型、动物模型或人肿瘤中具有肿瘤促进活性的任何哺乳动物基因,包括但不限于hras、kras、pik3ca、pik3cb、egfr、pdgfr、vegfr2、her2、src、syk、abl、raf或myc。在一些实施方案中,癌基因是来自下表1的基因之一。表1:文献中证实具有致癌或肿瘤促进活性的基因。在一些实施方案中,涉及肿瘤抑制基因的基因组修饰是切除肿瘤抑制基因或对肿瘤抑制基因活性关键的片段,其中所述基因(或其关键片段)先前已侧接重组酶位点,使得重组酶的表达导致切除肿瘤抑制基因或对肿瘤抑制基因活性关键的片段。重组酶位点可以是适于构建转基因动物的任何重组酶位点,诸如翻转酶(flp)、cre、dre、φc31整合酶、kd酵母重组酶、r酵母重组酶、b2酵母重组酶或b3酵母重组酶的重组酶位点。在一些实施方案中,涉及肿瘤抑制基因的基因组修饰是插入缺失(例如通过crisprsgrna定向的双链断裂)。在一些实施方案中,涉及肿瘤抑制基因的基因组修饰是并入插入缺失(例如通过crispr定向的双链断裂),该插入缺失引入了移码突变,其引起肿瘤抑制基因的提前终止或来自肿瘤抑制基因的开放阅读框的无义序列的表达。在一些实施方案中,涉及肿瘤抑制基因的基因组修饰是并入插入缺失(例如通过crisprsgrna定向的双链断裂),该插入缺失防止肿瘤抑制基因的转录(例如通过破坏肿瘤抑制启动子的关键元件)。在一些实施方案中,涉及肿瘤抑制基因的基因组修饰(即并入插入缺失)还涉及并入针对插入缺失起点的sgrna。在一些实施方案中sgrna伴有标识sgrna的条形码核酸序列(例如,以标识所靶向的肿瘤抑制基因中的特定位点,或标识所靶向的肿瘤抑制基因)。在一些实施方案中,sgrna伴有标识sgrna的条形码核酸序列和标识引入细胞中的单独dna分子的唯一分子标识符序列(umi)(例如,以标识单个肿瘤)。无论涉及肿瘤抑制基因的基因组修饰是重组酶介导的切除还是引入插入缺失到肿瘤抑制基因中,该肿瘤抑制基因都可以是证明在细胞模型、动物模型或人肿瘤中在部分或完全丧失功能的情况下具有肿瘤促进活性的任何哺乳动物基因,包括但不限于p53、lkb1、setd2、rb1、pten、nf1、nf2、tsc1、rnf43、ptprd、fbxw7、fat1、lrp1b、rasa1、lats1、arhgap35、ncoa6、ncor1、smad4、keap、ubr5、mga、clc、atf7ip、gata3、rbm10、cmtr2、arid1a、arid1b、arid2、smarca4、dnmt3、tet2、kdm6a、kmt2c、kmt2d、dot1l、ep300、atrx、brca2、bap1、ercc4、pole、atm、wm、cdkn2a、cdkn2c或stag2。在一些实施方案中,肿瘤抑制基因是来自下表2的基因之一。表2:文献中证实在部分或完全丧失功能的情况下具有肿瘤促进活性的基因。如上文所述,来自具有条形码的核酸的rna(例如,指导rna)和/或蛋白(例如,cre、crispr/casrna指导的蛋白等)的表达可以产生一个或多个基因组改变,并且在一些情况下,基因组改变导致靶细胞转化至赘生性细胞中(例如,在一些情况下,它可以导致肿瘤形成)。在一些实施方案中,靶细胞的基因组改变可以与赘生物特征的启动(例如,与肿瘤启动)在时间上分离。作为一个实例,一个或多个载体可以被工程改造为允许对crispr/cas指导rna的时间控制和/或对crispr/cas核酸指导的蛋白活性(例如,cas9活性)的时间控制。在一些情况下,引入遗传(例如,基因组)修饰的蛋白在靶细胞中表达。蛋白可以以蛋白或以编码蛋白的核酸(rna或dna)引入靶细胞中。蛋白也可以已经由细胞中的核酸编码(例如,由细胞中的基因组dna编码),并且方法包括诱导蛋白的表达。在一些情况下,将遗传修饰引入靶组织的靶细胞中的蛋白是基因组编辑蛋白/核酸内切酶(它们中的一些是“可编程的”,而另一些则不是“可编程的”)。实例包括但不限于:可编程的基因编辑蛋白(例如,转录激活子样(tal)效应物(tale)、tale核酸酶(talen)、锌指蛋白(zfp)、锌指核酸酶(zfn)、dna指导的多肽诸如格氏嗜盐碱杆菌(natronobacteriumgregoryi)argonaute(ngago)、crispr/casrna指导的蛋白(诸如cas9、casx、casy、cpf1)等等)(参见例如shmakov等人,natrevmicrobiol.2017年3月;15(3):169-182;和burstein等人,nature.2017年2月9日;542(7640):237-241);转座子(例如,i类或ii类转座子—如piggybac、睡美人(sleepingbeauty)、tc1/水手(tc1/mariner)、tol2、pif/先驱者(pif/harbinger)、hat、增变基因(mutator)、merlin、transib、helitron、maverick、青蛙王子(frogprince)、minos、himar1等等);大范围核酸酶(例如,i-scei、i-ceui、i-crei、i-dmoi、i-chui、i-diri、i-flmui、i-flmuii、i-anil、i-sceiv、i-csmi、i-pani、i-panii、i-panmi、i-sceii、i-ppoi、i-sceiii、i-ltri、i-gpii、i-gzei、i-onui、i-hjemi、i-msoi、i-tevi、i-tevii、i-teviii、pi-mlei、pi-mtui、pi-pspi、pi-tlii、pi-tliii、pi-scev等等);megatal(参见例如boissel等人,nucleicacidsres.2014年2月;42(4):2591–2601);噬菌体来源的整合酶;cre蛋白;flp蛋白;等等。在一些情况下,基因组编辑核酸酶(例如,crispr/casrna指导的蛋白)具有消除核酸酶活性(是核酸酶死亡蛋白)的一个或多个突变,并且该蛋白融合至转录激活子或阻遏子多肽(例如,crispra/crispri)。在一些情况下,基因组编辑核酸酶(例如,crispr/casrna指导的蛋白)具有消除核酸酶活性(是核酸酶死亡蛋白)或部分消除核酸酶活性(是切口酶蛋白)的一个或多个突变,可以具有调节蛋白功能或活性的一个或多个另外的突变,并且该蛋白融合至脱氨酶结构域(例如,adar、apobec1等),它本身可以具有调节蛋白功能或活性的一个或多个另外的突变,或融合至脱氨酶结构域和一个或多个另外的蛋白或肽(例如,噬菌体gam蛋白、尿嘧啶糖基化酶抑制剂等),它还可以具有调节蛋白功能或活性(例如,rna碱基编辑子、dna碱基编辑子)的一个或多个另外的突变。在一些情况下,编辑蛋白(诸如cre或flp)可以引入靶组织中,以用于诱导来自基因组的另一种蛋白(例如,crispr/casrna指导的蛋白(诸如cas9))的表达的目的,例如动物可以含有cas9的lox-stop-lox等位基因,并且所引入的cre蛋白(例如,由具有条形码的核酸编码)导致‘stop’的移除,从而引起cas9蛋白的表达。在一些实施方案中,具有条形码的核酸可以诱导赘生性细胞形成,并且包含以下各项中的一项或多项:同源定向修复(hdr)dna供体模板、编码癌基因的核酸(包括蛋白的野生型和/或突变型等位基因)、编码crispr/cas指导rna的核酸、编码短发夹rna(shrna)的核酸和编码基因组编辑蛋白的核酸(例如,参见上文)。在一些情况下,当具有条形码的核酸是hdrdna供体模板时,它们可以将突变引入靶细胞的基因组中。在一些此类情况下,基因组编辑核酸酶存在于将切割被靶向的dna从而将供体模板用于插入具有条形码的序列的细胞中(被引入或作为主题方法的一部分诱导或者已经在靶细胞中表达)。在一些情况下,hdrdna供体模板的文库(多个模板)包括具有对于每个分子的唯一序列标识符(条形码)的成员,但是这些分子产生相同的功能干扰(例如,它们都可以引起相同的蛋白(例如,在一些情况下具有突变的氨基酸序列)的表达,但是它们在密码子的摇摆位置中可以是不同的,然后编码蛋白,以使得所得的多个细胞谱系是可彼此区分的,尽管它们表达相同的突变蛋白)。在一些情况下,hdrdna供体模板的文库(多个模板)包括具有对于每个分子的唯一序列标识符(条形码)的成员,并且这些分子产生不同的功能干扰(例如,可以靶向不同的遗传基因座,可以靶向相同的基因座,但是引入不同的等位基因等)。在一些情况下,具有条形码的核酸是crispr/cas指导rna或是编码crispr/cas指导rna的dna分子。此类分子的文库可以包括靶向不同的基因座的分子和/或靶向相同的基因座的分子。在一些情况下,具有条形码的核酸编码癌基因,出于本公开的目的,该癌基因包括当过表达时可以导致赘生性细胞形成的野生型蛋白,以及可以导致赘生性细胞形成的突变蛋白(例如,kras–参见以下工作实例)。此类分子的文库可以包括表达相同的癌基因的分子或表达不同的癌基因的分子的文库。在一些情况下,具有条形码的核酸包括短发夹rna(shrna)和/或编码shrna的一种或多种dna分子(例如,这些核酸可以靶向任何所期望的基因,例如肿瘤抑制物)。此类分子的文库可以包括表达相同的shrna的分子或表达不同的shrna的分子的文库。在一些情况下,具有条形码的核酸包括编码一个或多个基因组编辑蛋白/核酸内切酶的rna和/或dna(参见上文,例如crispr/casrna指导的蛋白(诸如cas9、cpf1、casx或casy);cre重组酶;flp重组酶;zfn;talen;等等)。此类分子的文库可以包括表达相同的基因组编辑蛋白/核酸内切酶的分子或表达不同的基因组编辑蛋白/核酸内切酶的分子的文库。在一些实施方案中,细胞标志物是可区分标记的颗粒(例如,珠粒、纳米颗粒等等)。例如,在一些情况下,可以用可区分的质量标签(它可以经由质谱来分析)、用可区分的荧光蛋白、用可区分的放射性标签等等来标记颗粒。检测/测量/计算主题方法还可以包括例如在对于至少一部分可遗传标记的细胞经历至少一轮分裂而言已过去足够的时间之后,检测和测量所接触的组织中存在的多种细胞标志物中的至少两种的数量的步骤。在一些情况下,步骤(a)和(b)之间[使组织与多种细胞标志物接触以及检测/测量组织中存在的细胞标志物之间]经过的时期是足以使至少一部分可遗传标记的细胞(例如,至少两种可区分标记的细胞)经历至少一轮分裂(例如,至少2轮、4轮、6轮、8轮、10轮或15轮细胞分裂)的时期。在一些情况下,步骤(a)和(b)之间[使组织与多种细胞标志物接触以及检测/测量组织中存在的细胞标志物之间]经过的时期为2小时或更长(例如,4小时或更长、6小时或更长、8小时或更长、10小时或更长、12小时或更长、15小时或更长、18小时或更长、24小时或更长或36小时或更长)。在一些情况下,步骤(a)和(b)之间[使组织与多种细胞标志物接触以及检测/测量组织中存在的细胞标志物之间]经过的时期为1天或更多天(例如,2天或更多天、3天或更多天、4天或更多天、5天或更多天、7天或更多天、10天或更多天、或者15天或更多天、20天或更多天或者24天或更多天)。在一些情况下,步骤(a)和(b)之间[使组织与多种细胞标志物接触以及检测/测量组织中存在的细胞标志物之间]经过的时期为1周或更多周(例如,2周或更多周、3周或更多周、4周或更多周、5周或更多周、7周或更多周或者10周或更多周)。在一些情况下,步骤(a)和(b)之间[使组织与多种细胞标志物接触以及检测/测量组织中存在的细胞标志物之间]经过的时期在2小时至60周(例如,2小时至40周、2小时至30周、2小时至20周、2小时至15周、10小时至60周、10小时至40周、10小时至30周、10小时至20周、10小时至15周、18小时至60周、18小时至40周、18小时至30周、18小时至20周、18小时至15周、1天至60周、1天至40周、1天至30周、1天至20周、1天至15周、3天至60周、3天至40周、3天至30周、3天至20周、3天至15周、1周至60周、1周至40周、1周至30周、1周至20周或者1周至15周)的范围内。在一些情况下,步骤(a)和(b)之间[使组织与多种细胞标志物接触以及检测/测量组织中存在的细胞标志物之间]经过的时期在2小时至300周(例如,2小时至250周、2小时至200周、2小时至150周、2小时至100周、2小时至60周、2小时至40周、2小时至30周、2小时至20周、2小时至15周、10小时至300周、10小时至250周、10小时至200周、10小时至150周、10小时至100周、10小时至60周、10小时至40周、10小时至30周、10小时至20周、10小时至15周、18小时至300周、18小时至250周、18小时至200周、18小时至150周、18小时至100周、18小时至60周、18小时至40周、18小时至30周、18小时至20周、18小时至15周、1天至300周、1天至250周、1天至200周、1天至150周、1天至100周、1天至60周、1天至40周、1天至30周、1天至20周、1天至15周、3天至300周、3天至250周、3天至200周、3天至150周、3天至100周、3天至60周、3天至40周、3天至30周、3天至20周、3天至15周、1周至300周、1周至250周、1周至200周、1周至150周、1周至100周、1周至60周、1周至40周、1周至30周、1周至20周或者1周至15周)的范围内。对于每种可区分的细胞标志物(例如,具有条形码的核酸)所检测的信号的量(水平)可以用于确定所接触的组织(可遗传的细胞标志物所引入的组织)中存在的细胞的数量。任何方便的方法均可用于检测/测量细胞标志物,并且本领域的普通技术人员将理解,所用的细胞标志物的类型将驱动应使用哪种方法进行测量。例如,如果使用质量标签,则质谱分析可以是用于测量的所选方法。如果使用具有条形码的核酸作为细胞标志物,则测序(例如,高通量/下一代测序)可以是用于测量的所选方法。在一些情况下,使用高通量测序,并且对于每个所检测的条形码的序列读段数可以用于确定含有该特定条形码的细胞的数量。在一些情况下,重要性指标不是每个谱系中的细胞数量,而是超过一定数量细胞的克隆谱系的数量。在一些情况下,对pcr产物进行测序(例如,高通量/下一代测序),其中pcr产物来自从细胞内的细胞标志物(在一些情况下,从其中具有条形码的核酸整合的基因组区域)扩增条形码区域的pcr反应(参见例如图1a)。在一些情况下,肿瘤中的赘生性细胞的数量定量,以及另外的表型分型和分析,从合并的样品,经由单个、多个或组合排列的生物标志物(例如,荧光蛋白、细胞表面蛋白和抗体)分选的样品,或者经由来自组织、器官、细胞培养物或其他可能的细胞复制方法的单独的肿瘤的解剖来进行。在一些情况下,‘基准’可以用于帮助计算细胞数量。例如,在一些情况下,对照可以“加标(spike)”到样品中。例如,加标(掺标(spikein))对照可以用于确定每个细胞的序列读段数(例如,每个序列读段的细胞数)。例如,在一些情况下,加标(掺标)对照还可以用于将所测量的dna量与dna来源的细胞的数量相关联。例如,已知的细胞数可以用于制备dna,该dna可以与从所接触的组织(根据本公开的方法与可遗传的细胞标志物接触的组织)的细胞提取的dna平行处理。这种加标(掺标)对照(“基准”)可以包含其自身的唯一条形码。加标对照的结果可以用于推导/计算以测序反应中检测到的序列读段数表示的细胞数(即,加标(掺标)对照可以用于提供将测量值的量(例如,序列读段数)转换为细胞数量(例如,绝对细胞数)的系数)。这种过程可以称为“归一化”,例如测序结果提供了对于所检测的每个唯一条形码的读段数,然后可以将该值与一个或多个“基准”进行比较,以计算已包含所检测的唯一条形码的细胞的绝对数(参见例如图1a)。在一些情况下,因为可通过使主题组织与可遗传的细胞标志物接触来检测多个克隆细胞群,并且在一些情况下,每个可区分的细胞群具有类似的基因型,所以主题方法可以用于提供对于特定表型的群体大小的分布(例如,肿瘤大小的分布)。例如,如果初始接触在所有接触的细胞中产生类似的基因组改变(例如,如果所有细胞都接受靶向相同的基因座的指导rna,如果所有细胞都接受编码相同的癌基因等位基因的核酸等等),但是每个细胞群(例如,肿瘤)是独立的,则所得的细胞群大小可以提供对于特定基因型的克隆细胞群大小分布。例如,进行主题方法的目标可以是搜索以特定方式改变肿瘤行为的遗传变化(例如,改变大小分布,而不改变肿瘤本身的数量)。例如,以下工作实例(例如,参见工作实例1)包括这样的展示:携带具有p53缺陷型肿瘤的动物产生了对于最大肿瘤的幂律分布的肿瘤大小分布(与马尔可夫(markov)过程一致,其中非常大的肿瘤由另外的很少获得的驱动子突变产生)。相反,携带具有lkb1失活的肿瘤的动物增加大多数病灶的大小,这表明了正常的指数生长过程(例如,参见图10、13、16和20)。大小分布测量可以以多种不同方式使用。例如,可以通过进行本文所述的方法来测定对于给定基因型的细胞群大小(例如,肿瘤大小)的基线大小分布,并将其与当用测试化合物(例如,候选抗癌疗法)另外处理经类似处理的动物时测量的大小分布进行比较。大小分布的变化可以用作测试化合物是否有效的度量。作为一个示例性实例,本发明人测定了对于携带具有p53缺陷的肿瘤的小鼠的肿瘤大小分布的基线测量值,并且发现p53缺陷往往导致比其他肿瘤更大的一些肿瘤。因此,p53缺陷型肿瘤的大小分布不是标准分布,但包括异常肿瘤。使用本文所述的方法,可以筛选改变肿瘤大小分布,但是不一定治愈肿瘤动物的潜在疗法(例如,小分子、大分子、放疗、化疗、禁食、抗体、免疫细胞疗法、酶、病毒、生物制剂、化合物等等)。例如,可以发现这样的疗法(例如,化合物):虽然该疗法不能根除所有p53缺陷型肿瘤,但是它抑制了异常大的肿瘤的形成。使用标准方法可能无法检测到这种变化,因为测试化合物不一定使肿瘤总数(肿瘤负荷)减少或甚至使平均肿瘤大小减小(使用其他方法可能会丢弃这种化合物,因为化合物对抑制肿瘤生长无效)–但是这种疗法(例如,化合物)在临床环境中可以对治疗具有p53缺陷型肿瘤的患者非常有用,因为它将对最晚期肿瘤(例如,最大、更危险的肿瘤)有效(例如,减小异常肿瘤的风险)。因此,在一些情况下,主题方法可以用于筛选候选疗法(例如,小分子、大分子、放疗、化疗、禁食、抗体、免疫细胞疗法、酶、病毒、生物制剂、化合物等等)对群体大小的作用(例如,肿瘤的生长/增殖)。例如,主题方法可以在存在测试疗法(例如,化合物(例如,药物))的情况下进行(例如,方法可以包含使组织与测试化合物接触(例如,经由向个体的施用)的步骤),并且可以测量药物的作用,例如经由与其中未添加药物(例如,对照媒介物)的平行实验的比较。在其中谱系标记的细胞群在遗传上是相同的(或类似的)情况下,这种方法可以测试化合物是否对细胞群的大小分布具有作用。在其中差异标记的细胞具有不同的基因型(例如,不同的基因已突变和/或在不同的细胞谱系中表达)的情况下,可以同时针对多个不同的基因型来测试疗法(例如,化合物),例如在相同的动物中,在组织在活动物体内的情况下。i在一些情况下,这些实验和/或疗法(例如,化合物)筛选可以对培养物中生长的组织(例如,2d培养组织、3d培养组织、类器官培养物)进行。在一些情况下,这些方法可以在非人动物(诸如啮齿动物(例如,小鼠、大鼠)、猪、豚鼠、非人灵长类动物等等)中进行。可以评估任何干扰原(例如,小分子、大分子[例如,抗体或诱饵受体]、放疗、化疗、炎症诱导物、激素、纳米颗粒、免疫细胞疗法、酶、病毒、环境干预(例如,间歇性禁食、剧烈运动、饮食控制)等等)对多个标记的细胞群的群体大小的作用。遗传干扰也可以在所有克隆谱系中诱导,以评估它们的影响。在其中所有谱系都为相同的初始基因型的谱系的情况下,可以确定单独的克隆谱系(例如,肿瘤)的响应。在其中已诱导克隆谱系具有不同的确定改变的情况下,可以确定可诱导的遗传干扰对具有不同的改变的克隆谱系的影响。产生可诱导遗传改变的系统包括但不限于flp/frt或cre/loxp系统(在未用flp或cre调控的等位基因启动的细胞谱系中)或四环素可调控的系统(例如,具有一种或多种tre-cdna和/或一种或多种tre-shrna和/或一种或多种tre-sgrna的tta或rtta)的使用。可调控的crispr/cas9基因组编辑和赘生性细胞的二次转导可以在时间上产生基因组改变。在一些情况下,将在测量细胞标志物之前、期间和/或之后评估具有多个标记的细胞群的个体对外部干扰原(例如,候选抗癌疗法)的作用和响应(例如,药理学、化学、代谢、药代动力学、免疫原性、毒理学、行为作用和响应等)。在一些实施方案中,主题方法包括在产生可遗传标记的细胞(例如,可遗传标记的肿瘤)之后,将标记的细胞群中的一个或多个(例如,一个或多个肿瘤的全部或一部分)移植到接受者(例如,二次接受者)或多个接受者中,例如,从而将肿瘤接种到一个或多个接受者中。在一些情况下,这种步骤可以被视为类似于‘印影接种(replicaplating)’,其中可以针对测试化合物筛选大量动物,其中每只动物均从来自相同的起始肿瘤的细胞接种。因此,在一些情况下,方法包括其中将测试化合物施用于移植的一个或多个接受者的步骤(例如,方法可以包括检测和测量二次接受者中存在的多种细胞标志物中的至少两种的数量),例如以评估所移植的细胞的生长(并且在一些情况下,这可以在存在和/或不存在测试化合物的情况下进行)。因此,主题方法可以用作系列移植研究的一部分,其中最初产生的可遗传标记的细胞(例如,可遗传标记的肿瘤)被移植到一个或多个接受者中,并且可以对于可遗传标记的细胞的至少两种可区分的谱系来计算所接触的组织中存在的可遗传标记的细胞的数量。在上述一些情况下(例如,系列移植),测试化合物可以施用于系列移植接受者,并且可以将结果与对照(例如,接受移植但未接受测试化合物的动物、接受测试化合物但未接受移植的动物等等)进行比较。在一些实施方案中,一种或多种可遗传标记的细胞被重新标记(例如,重新编制条形码)。换句话说,在一些情况下,将已经过可遗传标记的细胞群体(例如,肿瘤)与第二多种细胞标志物接触,所述第二多种细胞标志物是可遗传且可彼此区分的,并且可与第一多种细胞标志物的细胞标志物区分。通过这种方式,使用者可以调查例如标记的细胞群(例如,肿瘤)内存在的差异。在某个实施方案中,可遗传标志物本身随时间推移而变化,从而用克隆谱系(例如,核苷酸条形码的进化)来记录细胞的系统发生。可遗传的谱系标志物也可以在表达的基因(内源性的或经工程改造的)内编码,该基因有利于通过来自标记的细胞的mrna或cdna的分析来确定细胞谱系。在一些情况下,细胞标志物被转换为不同类型的细胞标志物(例如,具有条形码的dna由标记的细胞表达为具有条形码的rna或蛋白)。在这种情况下,本领域的普通技术人员将理解,用于测量细胞标志物的方法将在测量时由期望测量的细胞标志物的类型来确定。例如,如果具有条形码的dna被用作细胞标志物,并且具有条形码的dna被表达为具有条形码的rna,则rna测序(例如,全转录物组测序、单细胞rna测序等)可以是用于测量rna条形码是否为期望测量的细胞标志物的类型的方法,或dna测序(例如,全基因组测序、全外显子组测序、被靶向的dna测序等)可以是用于测量dna条形码是否为期望测量的细胞标志物的类型的方法。在这种情况下,待测量的细胞标志物的选择可以由研究和直接连接至细胞标志物的期望的细胞表型驱动(例如,可以使用单细胞rna测序来测量具有条形码的rna细胞标志物,所以rna表达模式可以与细胞标志物直接连接)。在一些情况下,可以使用单细胞分析方法(例如,单细胞rna-seq、流式细胞术、质谱分析(cytof)、merfish、单细胞蛋白组分析)来测量细胞谱系标志物,以使得来自每个谱系的单独的细胞可以与来自每个另外谱系的单独的细胞相关联。在这种情况下,研究每个谱系内的细胞的表型。在这种情况下,这些分析也可以用于评估不同谱系的细胞对外部干扰(例如,药物处理)的表型响应。当检测和测量可遗传的细胞标志物(例如,具有条形码的核酸)时,在一些情况下,测量值来源于整个组织。因此,组织样品可以是从组织采集的部分,或可以是整个组织(例如,全肺、肾、脾、血液、胰腺等)。因此,细胞标志物(例如,核酸)可以从组织样品提取,以表示剩余的组织,或可以从整个组织提取。在一些情况下,生物学样品是血液样品。在一些情况下,生物学样品是血液样品,但是所接触的组织不是血液。例如,在一些情况下,可遗传标记的细胞可以将化合物(例如,唯一的分泌标志物,诸如蛋白或核酸)分泌至血液中,并且血液中存在的化合物的量可以用于计算存在所分泌的特定化合物的细胞的数量。例如,可遗传标记的细胞可以在一些情况下将荧光蛋白分泌至血液中,并且荧光蛋白可以被检测和测量并用于计算对于分泌该特定化合物的细胞的细胞群大小。在一些情况下,在未受干扰的个体中或在施用外部干扰原(例如,药物)后检测这些分泌的可遗传标志物。在一些情况下,生物学样品是体液(例如,血液、血浆、血清、尿液、唾液、来自腹膜腔的流体、来自胸膜腔的流体、脑脊液等)。在一些情况下,生物学样品是体液,但是所接触的组织不是体液。例如,在一些情况下,可遗传标记的细胞可以将分析物(例如,唯一标志物,诸如蛋白、核酸或代谢物)释放至尿液中,并且尿液中存在的化合物的量可以用于计算单独或响应于外部干扰原(例如,候选抗癌疗法)而释放该特定化合物的细胞的数量或细胞谱系的数量。在一些情况下,生物学样品中的细胞标志物的测量与可以与细胞标志物直接或间接相关的并且可以存在于相同的生物学样品中或单独的生物学样品中的细胞、细胞组分(例如,无细胞dna、rna、蛋白、代谢物等)或任何其他分析物(例如,dna、rna、蛋白、代谢物、激素、溶解氧、溶解二氧化碳、维生素d、葡萄糖、胰岛素、温度、ph、钠、钾、氯化物、钙、胆固醇、红细胞、血细胞比容、血红蛋白等)的分析平行进行。在一些情况下,如上文所述,检测和测量对从个体收集的生物学样品(例如,血液样品)进行。在一些情况下,检测和测量对所接触的组织的组织样品进行,所述组织样品在一些情况下可以是所接触的组织的一部分或可以是整个组织。当检测和测量时,可以考虑生物标志物(除所引入的可遗传细胞标志物之外)。例如,主题方法可以包括检测和/或测量可遗传标记的细胞的生物标志物,以及根据生物标志物测量的结果对可遗传标记的细胞进行分类的步骤。这种生物标志物可以表示任何数量的细胞特征,例如增殖状态(例如,ki-67蛋白的检测、brdu掺入等)、细胞类型(例如,使用各种细胞类型的生物标志物)、发育细胞谱系、干细胞特性(例如,细胞是否为干细胞和/或干细胞的类型)、细胞死亡(例如,膜联蛋白v染色、经切割的半胱天冬酶3、tunel等)和细胞信号传导状态(例如,检测信号传导蛋白的磷酸化状态,例如使用磷酸特异性抗体)。在一些情况下,理解某些疗法或干扰的基因型特异性可以用于告知(通过与其他疗法或干扰的相似性)该疗法或干扰的作用机制。通过发现基因型特异性,本文所公开的方法可以用于作出和测试对于确定基因型的组合疗法预测。可以对疗法组进行测试以建立其基因型特异性试剂盒和系统还提供了例如用于实践任何上述方法的试剂盒和系统。主题试剂盒和/或系统的内容物可能会有很大差异。试剂盒和/或系统可以包括例如以下各项中的一项或多项:(i)可彼此区分的可遗传细胞标志物(例如,具有条形码的核酸)的文库;(ii)用于执行主题方法的指示;(iii)用于从主题方法的检测和测量步骤所产生的值计算细胞数的软件;(iv)已配置的计算机系统。除上述组件之外,主题试剂盒还可以包括用于实践主题方法的说明。这些说明可以以各种形式存在于主题试剂盒中,这些形式中的一种或多种可以存在于试剂盒中。这些说明可能存在的一种形式是作为合适的介质或基质上的印刷信息(例如,上面印刷有信息的一张或多张纸)、在试剂盒的包装中、在包装插页中等。另一种方式是上面记录有信息的计算机可读介质,例如软盘、cd、闪存驱动器等。可能存在的另一种方式是网址,所述网址可以经由因特网使用,以访问已移除站点的信息。任何方便的方式都可以存在于试剂盒中。应用本公开的主题的各种应用的实例包括但不限于以下:定量更复杂的基因型的作用:本发明人已经产生和验证了表达crispr/cas单指导rna(sgrna)对,以便有利于每个肿瘤中两个靶基因的缺失的慢病毒载体。具有靶向肿瘤抑制物的成对组合的sgrna的慢病毒-cre载体的产生将会以高度平行方式发现肿瘤抑制物之间的协同和拮抗相互作用。具有靶向肿瘤抑制物的成对组合的sgrna的慢病毒-cre载体的产生将会以高度平行方式发现肿瘤抑制物之间的协同和拮抗相互作用。允许进行组合疗法筛选的多重体内基因组编辑。由于很多系统的适应能力,组合疗法正在成为治疗多种疾病的有效方式。潜在疗法组合的数量之多迅速产生了一种困难情况,在这种情况下,永远不能在患者中或甚至在临床前动物模型中测试每种组合。然而,药物组合的这种无法测试的基质可以含有对患者有效的组合。药物处理与编码另外的药物靶标的基因的crispr/cas介导的缺失组合可以允许对疗法组合进行多重建模。可以使用本公开中描述的组合物和方法来平行进行>100种成对药物处理的作用查询。例如,在人肺癌的小鼠模型中在三种肺癌基因型的背景下研究这些置换,将产生查询体内成对药物靶向的作用的半高通量系统。向其他癌症类型的扩展:本文所述的方法可以用于发现细胞生长/增殖的药物基因组敏感性(例如,在赘生物(例如,肺部腺癌)的情形中),并且所述方法可以应用于其中可区分的谱系的群体大小是所关注的任何方便的癌症类型和/或任何方便的情况。例如,本公开中概述的方法可以适用于可在基因工程模型(例如,肉瘤、膀胱癌、前列腺癌、卵巢癌、胰腺癌、造血癌等)中例如使用病毒载体诱导的任何癌症。鉴于人肺部腺癌中的肿瘤基因型的广泛多样性以及不断增加的潜在疗法的数量,本公开中描述的多重定量平台可以成为转化癌症生物学的支柱。本文所述的方法将允许使适当的疗法与适当的患者进行有效匹配的转化研究,并且将对临床患者护理产生直接影响。它还有助于用最可能对治疗产生响应的具有肿瘤的患者亚群进行临床试验-从而提高药物开发的成功率,并进一步拯救在靶向较差的临床试验中失败的药物。本公开的非限制性方面的实例上文所述的本发明的主题的方面(包括实施方案)单独或与一个或多个其他方面或实施方案组合可以是有利的。在不限制上述描述的情况下,下面提供了编号为1-57的本公开的某些非限制性方面。本领域的技术人员在阅读本公后时将显而易见的是,每个单独编号的方面可以与任何上述或下述单独编号的方面一起使用或组合。这旨在为所有这些方面的组合提供支持,并且不限于以下明确提供的方面的组合:1.一种测量相同的组织中的多个克隆细胞群的群体大小的方法,所述方法包括:(a)使生物学组织与可遗传且可彼此区分的多种细胞标志物接触,以在所接触的组织中产生可遗传标记的细胞的多种可区分的谱系;(b)在对于至少一部分所述可遗传标记的细胞经历至少一轮分裂而言已过去足够的时间之后,检测和测量所接触的组织中存在的所述多种细胞标志物中的至少两种的数量,从而产生一组测量值;以及(c)使用所述一组测量值作为输入来计算对于可遗传标记的细胞的至少两种所述可区分的谱系而言在所接触的组织中存在的可遗传标记的细胞的数量。2.根据1所述的方法,其中所接触的组织中的所述可遗传标记的细胞是赘生性细胞。3.根据1或2所述的方法,其中所述组织在步骤(a)之前包括赘生性细胞和/或肿瘤。4.根据1至3中任一项所述的方法,其中对从所述组织收集的生物学样品进行步骤(b)的所述检测和测量。5.根据1至3中任一项所述的方法,其中对所接触的组织的组织样品进行步骤(b)的所述检测和测量。6.根据1至5中任一项所述的方法,其中所述多种细胞标志物中的每种细胞标志物对应于可遗传标记的谱系的已知细胞基因型。7.根据1至6中任一项所述的方法,其中所述接触包括遗传改变所述组织的细胞以产生所述可遗传标记的细胞。8.根据1至7中任一项所述的方法,其中所述方法是测量相同的组织的多个肿瘤的肿瘤大小的方法。9.根据1至8中任一项所述的方法,其中接触所述组织的步骤包括诱导赘生性细胞。10.根据1至9中任一项所述的方法,其中所述细胞标志物是诱导或修饰赘生性细胞形成和/或肿瘤形成的剂。11.根据1至10中任一项所述的方法,其中所述检测和测量在对于由于所述接触而在所接触的组织中形成肿瘤而言已过去足够的时间之后进行。12.根据1至11中任一项所述的方法,其中所述多种细胞标志物包括具有条形码的核酸。13.根据12所述的方法,其中所述检测和测量包括对每个所检测的条形码的序列读段的高通量测序以及对所述读段的数量的定量。14.根据1至13中任一项所述的方法,其中所述多种细胞标志物包括诱导赘生性细胞形成的具有条形码的核酸。15.根据12至14中任一项所述的方法,其中所述具有条形码的核酸诱导赘生性细胞形成并且包括以下各项中的一项或多项:同源定向修复(hdr)dna供体模板、编码一种或多种癌基因的核酸、编码一种或多种野生型蛋白的核酸、编码一种或多种突变型蛋白的核酸、编码一种或多种crispr/cas指导rna的核酸、编码一种或多种短发夹rna(shrna)的核酸以及编码一种或多种基因组编辑蛋白的核酸。16.根据权利要求15所述的方法,其中所述基因组编辑蛋白选自:crispr/casrna指导的蛋白、融合至转录激活子或阻遏子多肽的crispr/casrna指导的蛋白、cas9蛋白、融合至转录激活子或阻遏子多肽的cas9蛋白、锌指核酸酶(zfn)、talen、噬菌体来源的整合酶、cre蛋白、flp蛋白和大范围核酸酶蛋白。17.根据12至16中任一项所述的方法,其中所述具有条形码的核酸是线性或环状dna分子。18.根据12至16中任一项所述的方法,其中所述具有条形码的核酸选自:质粒、合成核酸片段和小环。19.根据12至16中任一项所述的方法,其中所述具有条形码的核酸是rna分子。20.根据12至16中任一项所述的方法,其中所述具有条形码的核酸是rna/dna杂合体或核酸/蛋白质复合物。21.根据1至19中任一项所述的方法,其中所述组织是无脊椎动物组织。22.根据1至19中任一项所述的方法,其中所述组织是脊椎动物组织。23.根据1至19中任一项所述的方法,其中所述组织是哺乳动物或鱼组织。24.根据1至19中任一项所述的方法,其中所述组织是大鼠组织、小鼠组织、猪组织、非人灵长类动物组织或人组织。25.根据1至24中任一项所述的方法,其中所述组织是活动物的一部分。26.根据1至24中任一项所述的方法,其中所述组织是在动物体外生长的经工程改造的组织。27.根据1至26中任一项所述的方法,其中所述组织选自:肌肉、肺、支气管、胰腺、乳腺、肝、胆管、胆囊、肾、脾、血液、肠、脑、骨、膀胱、前列腺、卵巢、眼、鼻、舌、口、咽、喉、甲状腺、脂肪、食管、胃、小肠、结肠、直肠、肾上腺、软组织、平滑肌、脉管系统、软骨、淋巴、前列腺、心脏、皮肤、视网膜、繁殖系统和生殖系统。28.根据1至27中任一项所述的方法,其中在对于至少一部分所述可遗传标记的细胞经历至少一轮分裂而言已过去足够的时间之后,所述方法还包括:(i)检测和/或测量所述可遗传标记的细胞的生物标志物,以及(ii)根据所述生物标志物的所述检测和/或测量的结果对所述可遗传标记的细胞进行分类。29.根据28所述的方法,其中所述生物标志物为以下各项中的一项或多项:细胞增殖状态、细胞类型、发育细胞谱系、细胞死亡和细胞信号传导状态。30.根据1至29中任一项所述的方法,其中所述细胞标志物经由病毒载体递送至所述组织。31.根据30所述的方法,其中所述病毒载体选自:慢病毒载体、腺病毒载体、腺相关病毒(aav)载体和逆转录病毒载体。32.一种测量相同的组织的多个克隆独立的肿瘤的肿瘤大小的方法,所述方法包括:(a)使组织与多个具有条形码的核酸细胞标志物接触,从而在所接触的组织中产生可遗传标记的赘生性细胞的多种可区分的谱系;(b)在对于至少一部分所述可遗传标记的赘生性细胞经历至少一轮分裂而言已过去足够的时间之后,进行高通量核酸测序以检测和测量所接触的组织中存在的所述具有条形码的核酸细胞标志物中的至少两种的数量,从而产生一组测量值;以及(c)使用所述一组测量值作为输入来计算对于可遗传标记的赘生性细胞的至少两种所述可区分的谱系而言在所接触的组织中存在的可遗传标记的赘生性细胞的数量。33.根据32所述的方法,其中所述组织在步骤(a)之前包括赘生性细胞和/或肿瘤。34.根据32或33所述的方法,其中对从所述组织收集的生物学样品进行步骤(b)的所述高通量核酸测序。35.根据32或33所述的方法,其中对所接触的组织的组织样品进行步骤(b)的所述高通量核酸测序。36.根据32至35中任一项所述的方法,其中所述多种具有条形码的核酸细胞标志物中的每种具有条形码的核酸细胞标志物对应于可遗传标记的赘生性细胞谱系的已知细胞基因型。37.根据32至36中任一项所述的方法,其中所述接触包括遗传改变所述组织的细胞以产生所述可遗传标记的赘生性细胞。38.根据32至37中任一项所述的方法,其中所述具有条形码的核酸诱导赘生性细胞形成。39.根据32至37中任一项所述的方法,其中所述具有条形码的核酸诱导赘生性细胞形成并且包括以下各项中的一项或多项:同源定向修复(hdr)dna供体模板、编码一种或多种癌基因的核酸、编码一种或多种野生型蛋白的核酸、编码一种或多种突变型蛋白的核酸、编码crispr/cas指导rna的核酸、编码短发夹rna(shrna)的核酸以及编码基因组编辑蛋白的核酸。40.根据39所述的方法,其中所述基因组编辑蛋白选自:crispr/casrna指导的蛋白、融合至转录激活子或阻遏子多肽的crispr/casrna指导的蛋白、cas9蛋白、融合至转录激活子或阻遏子多肽的cas9蛋白、锌指核酸酶(zfn)、talen、噬菌体来源的整合酶、cre蛋白、flp蛋白和大范围核酸酶蛋白。41.根据32至40中任一项所述的方法,其中所述具有条形码的核酸是线性或环状dna分子。42.根据32至40中任一项所述的方法,其中所述具有条形码的核酸选自:质粒、合成核酸片段和小环。43.根据32至42中任一项所述的方法,其中所述具有条形码的核酸是rna/dna杂合体或核酸/蛋白质复合物。44.根据32至43中任一项所述的方法,其中所述组织是无脊椎动物组织。45.根据32至43中任一项所述的方法,其中所述组织是脊椎动物组织。46.根据32至43中任一项所述的方法,其中所述组织是哺乳动物或鱼组织。47.根据32至43中任一项所述的方法,其中所述组织是大鼠组织、小鼠组织、猪组织、非人灵长类动物组织或人组织。48.根据32至47中任一项所述的方法,其中所述组织是活动物的一部分。49.根据32至47中任一项所述的方法,其中所述组织是无脊椎动物组织。在动物体外生长的经工程改造的组织。50.根据32至49中任一项所述的方法,其中所述组织选自:肌肉、肺、支气管、胰腺、乳腺、肝、胆管、胆囊、肾、脾、血液、肠、脑、骨、膀胱、前列腺、卵巢、眼、鼻、舌、口、咽、喉、甲状腺、脂肪、食管、胃、小肠、结肠、直肠、肾上腺、软组织、平滑肌、脉管系统、软骨、淋巴、前列腺、心脏、皮肤、视网膜和繁殖系统以及生殖系统。51.根据32至50中任一项所述的方法,其中在对于至少一部分所述可遗传标记的赘生性细胞经历至少一轮分裂而言已过去足够的时间之后,所述方法还包括:(i)检测和/或测量所述可遗传标记的赘生性细胞的生物标志物,以及(ii)根据所述生物标志物的所述检测和/或测量的结果对所述可遗传标记的赘生性细胞进行分类。52.根据51所述的方法,其中所述生物标志物为以下各项中的一项或多项:细胞增殖状态、细胞类型、发育细胞谱系、细胞死亡和细胞信号传导状态。53.根据32至52中任一项所述的方法,其中所述细胞标志物经由病毒载体递送至所述组织。54.根据53所述的方法,其中所述病毒载体选自:慢病毒载体、腺病毒载体、腺相关病毒(aav)载体、博卡病毒载体、泡沫病毒载体和逆转录病毒载体。55.根据1至54中任一项所述的方法,其中所述方法包括使所述组织与测试化合物(例如,测试药物)接触,以及确定所述测试化合物是否对细胞群体大小和/或细胞群体大小的分布具有作用。56.根据1至55中任一项所述的方法,其中在产生所述可遗传标记的细胞之后,所述方法包括将所述可遗传标记的细胞中的一者或多者移植(例如,移植一种或多种肿瘤)到一个或多个接受者(例如,二次接受者,例如以将肿瘤接种到所述二次接受者中)。57.根据56所述的方法,其中测试化合物施用于所述一个或多个接受者,并且所述方法包括检测和测量所述一个或多个接受者中的所述多种细胞标志物中的至少两种的数量(例如,以评估响应于所述测试化合物的存在的所述移植细胞的生长)。实施例给出以下实施例以便为本领域的普通技术人员提供如何制备和使用本发明的完整公开和描述,并且既不旨在限制发明人所认为的他们的发明的范围,也不旨在表示以下实验是所进行的全部或仅有的实验。已经作出了确保关于所用数字(例如,量、温度等)的准确性的努力,但是应考虑某些实验误差和偏差。除非另外说明,否则份数是重量份,分子量是重均分子量,温度是摄氏度,并且压力是大气压或接近大气压。实施例1:tuba-seq:发现体内肿瘤抑制的适合度景观的定量和多重方法癌症生长和进展是多阶段、随机的进化过程。虽然癌症基因组测序在鉴定人肿瘤中出现的基因组改变中发挥了重要作用,但是这些改变对天然组织内的肿瘤生长的效果仍然未得到充分的探索。人癌症的基因工程小鼠模型能够进行体内肿瘤生长研究,但是以精确和可调整的方式定量所得的肿瘤大小的方法的缺乏限制了我们理解单独的肿瘤抑制基因的作用大小和模式的能力。在这里,我们提出了一种将肿瘤条形码编制(tumorbarcoding)与超深度条形码测序(barcodesequencing)整合(tuba-seq)以在人癌症的小鼠模型中查询肿瘤抑制功能的方法。tuba-seq发现了肿瘤大小在肺部肿瘤的三个原型基因型中的不同分布。通过将tuba-seq与多重crispr/cas9介导的基因组编辑组合,我们还定量了十一个最经常失活的肿瘤抑制途径在人肺部腺癌中的作用。该方法将甲基转移酶setd2和剪接因子rbm10鉴定为肺部腺癌生长的新型抑制物。tuba-seq具有前所未有的分辨率、平行度和精度,能够进行肿瘤抑制基因功能的适合度景观的广泛定量。结果肿瘤条形码编制(tumorbarcoding)与超深度条形码测序(barcodesequencing)(tuba-seq)能够进行肿瘤大小的精确和平行定量。致癌性kras是人肺部腺癌的关键驱动因素,并且早期肺部肿瘤可以使用loxp-stop-loxpkrasg12d敲入小鼠(kraslsl-g12d/+)来建模,其中cre在肺上皮细胞中的表达引起致癌性krasg12d的表达。lkb1和p53是致癌性kras驱动的人肺部腺癌中的经常突变的肿瘤抑制物,并且lkb1缺陷和p53缺陷增加了致癌性krasg12d驱动的肺部肿瘤的小鼠模型中的肿瘤负荷(图7a)。病毒cre诱导的肺癌小鼠模型能够同时启动许多肿瘤,并且可以通过慢病毒介导的dna条形码编制来稳定标记单独的肿瘤。所以,我们试图确定来自成批携带肿瘤的肺的慢病毒条形码区的高通量测序是否可以定量每个具有唯一条形码的肿瘤中的癌细胞数(图7b)。为了查询致癌性krasg12d驱动的肺部肿瘤的生长以及lkb1和p53丧失对肿瘤生长的影响,我们用含有大于106个唯一dna条形码的慢病毒cre载体的文库(lenti-mbc/cre;图1a和图7b)在kraslsl-g12d/+;rosa26lsl-tomato(kt)、kt;lkb1flox/flox(klt)和kt;p53flox/flox(kpt)小鼠中启动肺部肿瘤。在肿瘤启动后十一周,kt小鼠产生广泛的增生和一些小肿瘤块(图1b和图7c)。有趣的是,klt小鼠具有大小相对均匀的大型肿瘤,kpt小鼠具有各种不同的肿瘤大小(图1b)。为了使用超深度测序来定量每个病灶中的癌细胞数,我们pcr扩增从每只小鼠分离的约1/10的成批肺dna的整合慢病毒条形码区并对其进行测序至大于107个读段/小鼠的平均深度(图1a,方法)。我们观察到小鼠中的超过一千倍的肿瘤大小差异(图1c)。来自小病灶的条形码读段可以表示唯一的肿瘤,或从来自较大肿瘤的类似的条形码的反复出现的测序误差产生。为了使这些假性肿瘤的发生最小化,我们使用产生测序误差的统计模型的算法来聚集预期来源于相同的肿瘤条形码的读段(dada2:图2和图8)。进一步选择dada2聚集率和最小肿瘤大小以使我们的肿瘤识别流程的再现性最大化(图8d-f)。这些方法极大地限制了反复出现的测序误差对肿瘤定量的作用,但可能不会完全消除该作用(图2a)。每个肿瘤中的绝对癌细胞数的定量将允许来自相同基因型的单独小鼠的数据的聚集以及基因型之间肿瘤大小的比较。为了使读段计数转换为癌细胞数,我们在组织匀浆和dna提取之前将具有已知条形码的细胞以确定的数量添加至每个肺部样品(图1a和图9)。因此,通过将肿瘤读段计数归一化为“基准”读段计数,我们可以计算每只小鼠中的每个肿瘤中的绝对癌细胞数(图1a和图9)。tuba-seq在技术重复之间具有高度可重现性,并且对可以使肿瘤大小分布产生偏差的很多技术变量不灵敏,这些变量包括测序误差、单个测序机器的内在误差率的差异、条形码gc含量、条形码多样性、小鼠中的肿瘤数量和读段深度(图2b-d,图10)。虽然适度的测量误差以较小的大小存在,但是这些不会使总体大小分布产生偏差。肿瘤大小分布在相同基因型的小鼠之间也具有高度可重现性(r2>0.98;图2e、f、图10g)。事实上,根据基因型的大小分布的无监督层次聚类明确地区分了小鼠,即使在用不同滴度的lenti-mbc/cre来诱导肿瘤时(图2g和图10d)。然而,我们的方法检测了相同基因型的小鼠之间的肿瘤大小谱差异。该差异远大于在相同小鼠中的两个肿瘤部分之间观察到的随机噪声,这表明tuba-seq比小鼠之间的肿瘤负荷的内在差异显著更精确(图2e、g)。因此,tuba-seq可快速和精确定量kt、klt和kpt小鼠中的几千个肺部病灶中的癌细胞数(图1c、图10c)。肿瘤大小的分析发现了两种肿瘤抑制模式为了评估p53缺陷或lkb1缺陷对肿瘤生长的作用,我们计算了分布内的不同百分位处的肿瘤中的癌细胞数。有趣的是,虽然klt小鼠中的肿瘤始终大于kt肿瘤,但是p53的缺失不会改变大多数肿瘤中的癌细胞数(图3a-c)。相反,一小部分p53缺陷型肿瘤生长至异常大小,并且在任何小鼠中是最大的(图1c)。为了更好地理解p53和lkb1缺陷所赋予的肿瘤生长差异,我们确定了最佳拟合kt、klt和kpt小鼠中的肿瘤大小分布的数学分布。lkb1缺陷型肿瘤在整个分布范围内呈对数正态分布(图3d)。预期对数正态分布来自以正态分布速率进行的简单指数肿瘤生长。为了在不允许非常大的肿瘤大大改变平均肿瘤大小的情况下估算该指标,我们还计算了给定肿瘤大小的对数正态分布的平均癌细胞数的最大似然估算值(ln平均值)。通过该测量发现,klt肿瘤的癌细胞比kt肿瘤平均多7倍,这与lkb1在抑制增殖中的作用一致(图3a、c)。虽然在kpt小鼠中肿瘤负荷更大且肿瘤明显更大,但是p53缺陷不会增加我们的平均病灶大小估算值。相反,p53缺陷型肿瘤在较大的大小处呈幂律分布,并且总体肿瘤负荷升高是由罕见、超大的肿瘤驱动的(图3d)。这表明p53缺陷型肿瘤带来了驱动后续快速生长的另外的罕见、但深刻的肿瘤发生事件。用于肿瘤抑制基因的多重crispr/cas9介导的失活的具有条形码的慢病毒载体的文库的产生人肺部腺癌具有多种基因组改变,但是描述它们对肿瘤生长的影响的定量数据却很少(图7a和12b)。为了同时平行定量很多已知和候选肿瘤抑制基因的肿瘤抑制功能,我们将tuba-seq和常规基于cre的小鼠模型与多重crispr/cas9介导的体内基因组编辑组合(图4a-c)。评估单只小鼠中的不同肿瘤基因型还应通过消除小鼠间差异的影响来使tuba-seq分辨率最大化。我们首先通过用靶向tdtomato报告基因或lkb1的慢病毒-sgrna/cre载体来启动肿瘤确认了具有h11lsl-cas9等位基因的小鼠中的肺部肿瘤中有效的cas9介导的基因失活(图11)。在大约40%的肿瘤中实现了tdtomato的纯合失活,并且cas9介导的lkb1失活增加了肿瘤负荷(图11)。这些数据表明我们能够使用这些方法来遗传改变kras驱动的肺癌模型中的肿瘤。我们选择了十一个已知和推定的肺部腺癌肿瘤抑制基因,它们表示多种途径,包括广泛参与染色质重塑(setd2和arid1a)、剪接(rbm10)、dna损伤响应(atm和p53)、细胞周期控制(rb1和cdkn2a)、营养和氧化应激感测(lkb1和keap1)、环境应激响应(p53)以及tgf-β和wnt信号传导(分别为smad4和apc)的基因(图4b和图7a)。我们鉴定在转录物的早期、已知功能结构域的上游和人肿瘤中存在的大多数突变的上游产生插入缺失的有效sgrna(图12a)。为了允许使用tuba-seq来准确定量每个肿瘤中的癌细胞数,我们对靶向每个肿瘤抑制物的lenti-sgrna/cre载体和四个具有双组分条形码的lenti-sginert/cre阴性对照载体进行了多样化处理。该条形码由每个sgrna特有的唯一的8个核苷酸的“sgid”和唯一标记每个肿瘤的随机的15个核苷酸的条形码(bc)(sgid-bc;图4a、b和图12c-e)组成。单独和在库中确定了针对每个sgrna的体外切割效率(图13)。体内肿瘤抑制功能的平行定量为了以多重方式定量每个基因的失活对肺部肿瘤生长的影响,我们用十一个具有条形码的lenti-sgrna/cre载体和四个具有条形码的lenti-sginert/cre载体的库在kt和kt;h11lsl-cas9(kt;cas9)小鼠中启动了肿瘤(lenti-sgts-pool/cre;图4b、c)。尽管与kt小鼠相比接受了较低剂量的病毒,但是在肿瘤启动后12周kt;cas9小鼠相对于kt小鼠而言宏观肿瘤数量和大小增加(图4d、e)。为了确定具有每个sgrna的每个肿瘤中的癌细胞数,我们扩增了携带肿瘤的成批肺dna的sgid-bc区,对产物进行了深度测序,并应用了我们的tuba-seq分析流程。我们计算了相对于每只小鼠中的无活性sgrna的分布的对于每个肿瘤抑制物的生长效应的整体分布。对于每个sgrna,用分布中的不同百分位的肿瘤中的癌细胞数除以无活性分布中的对应百分位的大小(图5a)。这种相对和小鼠内比较使tuba-seq的精度最大化(方法)。我们还确定了含有十一个靶向肿瘤抑制物的sgrna中的每个的肿瘤的相对对数正态(ln)平均大小,以鉴定通常抑制癌症生长的肿瘤抑制物(图5b)。这些分析确认了lkb1、rb1、cdkn2a和apc在krasg12d驱动的肺部肿瘤生长中的已知肿瘤抑制功能(图5a、b和图12b)。在kt小鼠(缺乏h11lsl-cas9等位基因)中用lenti-sgts-pool/cre启动的肿瘤在具有每个sgrna的肿瘤的大小分布方面仅具有很小的差异(图14a-c)。为了评估该方法的再现性,我们在用lenti-sgts-pool/cre进行肿瘤启动后15周分析了另外的kt;cas9小鼠队列。我们确认了在肿瘤启动后12周鉴定的所有肿瘤抑制物的肿瘤抑制作用(图5c和图14e-f)。我们使用多重慢病毒-sgrna/cre递送和肿瘤条形码测序来检测肿瘤抑制物的能力是可重现的,如第95个百分位的肿瘤的ln平均大小和相对癌细胞数二者所评估(图5c和图14e、f)。第95个百分位的肿瘤的生长效应是非常高度相关的(r2=0.953),并且在两个时间点之间的与ln平均值相关的p值是类似的,尽管在15周时间点仅使用3只小鼠(图5c)。p53介导的肿瘤抑制的鉴定和肿瘤抑制物库中的肿瘤大小分布的概述与kpt小鼠中的肿瘤大小分布一致的是,最多第95个百分位的肿瘤的ln平均值和分析二者都不能在具有lenti-sgtspool/cre启动的肿瘤的kt;cas9小鼠中发现靶向p53的作用(图5)。不出所料,lenti-sgp53/cre启动的肿瘤表现出在较大的大小处呈幂律分布,并且在具有lenti-sgtspool/cre诱导的肿瘤的kt;cas9小鼠中sgp53被富集在最大的肿瘤内(图15a、b)。这与p53失活一致,p53失活允许一小部分肿瘤生长到较大的大小。在随后的15周时间点,靶向p53的作用更大,这与另外的改变的逐步积累以及p53在限制肿瘤进展中的已知作用一致(图15a、图15b)。重要的是,在具有lenti-sgtspool/cre启动的肿瘤的kt;cas9小鼠中,lkb1缺陷型肿瘤表现出肿瘤大小的对数正态分布,这与来自klt小鼠的数据一致(图16a)。因此,通过crispr/cas9介导的基因组编辑产生的p53缺陷型和lkb1缺陷型肿瘤具有与使用传统的floxed等位基因启动的那些肿瘤类似的大小分布。这表明,即使在合并的情况下,单独的肿瘤大小的定量可以发现在肿瘤抑制物失活时的肿瘤大小的独特和特征性分布。作为体内肺部肿瘤生长的抑制物的setd2和rbm10的鉴定有趣的是,除了适当地发现对体内肺部肿瘤生长具有已知作用的若干肿瘤抑制物之外,tuba-seq还鉴定出作为肺部肿瘤生长的主要抑制物的甲基转移酶setd2和剪接因子rbm10。setd2是唯一的组蛋白h3k36me3甲基转移酶,而且还可以通过微管的甲基化来影响基因组稳定性。尽管setd2在若干主要癌症类型(包括肺部腺癌)中是经常突变的,但是关于它作为体内肿瘤抑制物的作用知之甚少。setd2失活大大增加了肿瘤大小,很多含有sgsetd2的肿瘤具有的癌细胞比对照肿瘤多超过五倍(图5a、b和图16b)。有趣的是,用lenti-sgsetd2/cre启动的肿瘤表现出肿瘤大小的对数正态分布(图16c)。事实上,只有lkb1失活产生了类似的适合度优点,这强调了setd2突变在肺部腺癌患者中驱动大量肿瘤生长的潜在重要性(图16)。在很多癌症类型中,剪接因子也已经成为潜在的肿瘤抑制物。虽然在10-15%的人肺部腺癌中,剪接体的组分是突变的,但是关于它们对肿瘤抑制的功能贡献知之甚少。rbm10失活显著增加了前50%的肺部肿瘤中的癌细胞数,并且增加了ln平均大小(图5a、b)。这些数据表明,setd2介导的赖氨酸甲基化和异常前体mrna剪接各自在肺部腺癌中具有深刻的促肿瘤发生作用。tuba-seq是定量体内肿瘤抑制的精确和灵敏的方法对相同的小鼠中具有独特遗传改变的很多肿瘤中的癌细胞数的定量允许鉴定和消除多个来源的生物学和技术差异(方法)。通过启动每只小鼠的多个病灶、对每个病灶进行条形码编制、将多个sgrna并入每只小鼠以及将inert的sgrna纳入库中,我们可以鉴定和校正肿瘤生长中的很多来源的差异。如果没有这些关键特征,我们的分析就会由于以下因素而失败:所启动的肿瘤数量的差异(cv=27%)、相同基因型的小鼠之间的平均肿瘤大小(cv=38%)以及使单独的小鼠中的不同肿瘤抑制基因失活的平均作用大小之间的微小相关性(cv=11%)。通过计算每个肿瘤的大小,而不是使用成批测量值(诸如所有肿瘤中的sgrna呈现),我们更精确和灵敏地确定了使不同的肿瘤抑制物失活的生长作用。有趣的是,我们鉴定的肿瘤抑制物(apc、rb1、rbm10和cdkn2a)的三分之二仅在我们考虑每个具有条形码的肿瘤中的癌细胞数时,而不是我们仅考虑sgid呈现的倍数变化时鉴定(图5d)。事实上,与仅分析sgid呈现的变化相比,使用tuba-seq流程使作用大小、统计学显著性以及检测具有较小作用的肿瘤抑制物的能力都得以改进(图5e、f)。因此,tuba-seq提供了准确捕获功能性肿瘤抑制基因的生长抑制作用所需的分辨率水平。中靶crispr/cas9介导的基因组编辑的确认作为研究针对肿瘤抑制物失活的选择以及确认中靶sgrna介导的基因组编辑的正交方法,我们对来自三只lenti-sgts-pool/cre感染(转导)的kt;cas9小鼠的成批肺dna的每个sgrna靶向区进行了pcr扩增和深度测序。相对大部分setd2、lkb1和rb1等位基因在靶位点具有失活的插入缺失,这与中靶sgrna活性和这些基因失活的肿瘤的扩增一致(图6a以及图15c-f和17a,b)。来自lenti-sgts-pool/cre感染(转导)的kt;cas9小鼠的成批肺dna的这些基因的靶向区的扩增和测序还确认所有靶基因都含有插入缺失(图6a)。虽然我们的库中包含的所有基因在人肺部腺癌中是反复突变的并且在具有致癌性kras的肿瘤中是经常突变的(图7a),但是未通过作为肿瘤抑制物的任何指标鉴定到arid1a、smad4、keap1和atm(图5和6a以及图14d-f)。atm的肿瘤抑制功能的缺乏与使用atmfloxed等位基因的结果一致,并且我们确认在用lenti-sgsmad4/cre感染(转导)的kt;cas9小鼠中smad4对致癌性krasg12d驱动的体内肺部肿瘤生长的肿瘤抑制功能缺乏(图17c、d)。对于这些基因,基因表达或环境状态的变化、另外的时间或者同时发生的癌基因和/或肿瘤抑制物改变对于这些途径的失活以赋予肺癌细胞生长优点可以是必需的。为了进一步验证setd2的肿瘤抑制作用以及评估setd2缺陷型肿瘤的组织学,我们在kt和kt;cas9小鼠中用含有无活性sgrna(sgneo2)或两个不同的靶向setd2的sgrna中的任一个的慢病毒载体诱导了肿瘤。具有用任一种lenti-sgsetd2/cre载体启动的肿瘤的kt;cas9小鼠产生较大的腺瘤和腺癌,并且与具有用相同的病毒启动的肿瘤的kt小鼠相比具有显著更大的总体肿瘤负荷(图6b、c)。虽然这些小鼠的组织学分析发现了较大的小鼠间差异,但是通过tuba-seq进行的单独的肿瘤大小分析确认,与对照肿瘤相比,在setd2缺陷型肿瘤中癌细胞数几乎增加四倍(图6c、d和图18)。重要的是,通过常规方法进行的setd2肿瘤抑制的验证与我们的十一个推定的肿瘤抑制物的初始筛选相比需要更多的小鼠,这强调了多重sgrna可增加通量和降低成本的有益效果。讨论虽然已经从癌症基因组测序鉴定了很多推定的肿瘤抑制物,但是以快速、系统和定量的方式测试它们的功能(例如,体内)的有限策略仍然存在(图19)。通过组合dna条形码编制、高通量测序和crispr/cas9介导的基因组编辑,tuba-seq不仅增加了这些分析的通量,而且还能够进行体内肿瘤生长的非常精确和详细的定量。有趣的是,在相同的时间、相同的小鼠中启动的具有相同的基因组改变的肿瘤在仅12周生长后即生长至完全不同的大小。因此,另外的自发改变、初始转化细胞的状态差异或局部微环境可以影响肿瘤如何迅速生长及其是否具有连续扩增的能力。tuba-seq还具有发现肿瘤大小的基因型特异性分布的独特能力,该分布揭示了基因功能的性质。p53缺陷产生对于最大的肿瘤呈幂律分布的肿瘤大小分布,这与马尔可夫过程一致,其中非常大的肿瘤由另外的很少获得的驱动子突变产生。相反,lkb1失活增加了大多数病灶的大小,这表明是正常的指数生长过程。因此,肿瘤抑制物可以具有经由tuba-seq鉴定的、可以预示它们的分子功能的不同的肿瘤抑制模式。有趣的是,最近表明setd2可以使微管蛋白甲基化,并且setd2缺陷可以产生各种形式的基因组不稳定性,包括由于微管的改变而产生的微核和染色体迟滞。预计基因组不稳定性会产生罕见、有利的改变以及高度随机和呈幂律分布的肿瘤生长。然而,在我们的研究中,setd2缺陷型肺部肿瘤的大小分布呈严格对数正态分布,所以我们推测setd2丧失的主要影响是诱导通常使生长失调的基因表达程序(图6d和图16b、c)。我们的分析规模(评估数千个单独的肿瘤)极大地提高了我们鉴定功能性肿瘤抑制基因的能力。经由成批测量进行的肿瘤生长估算仅鉴定出我们发现对肿瘤生长有利的肿瘤抑制物的三分之一(图5d-f)。与常规floxed等位基因不同的是,在肺中crispr/cas9介导的基因组编辑在所有肿瘤的大约一半中产生了纯合的无效等位基因(图11d)。因此,虽然靶基因的一致纯合缺失的缺乏将减少来自成批测量的肿瘤抑制性信号,但是通过对每个肿瘤进行条形码编制和分析,tuba-seq有效地克服了这一技术局限性。通过分析大量肿瘤抑制物,我们的数据表明早期赘生性细胞处于进化新生状态,其中很多肿瘤抑制物改变是适应性的,并具有生长优点。相比之下,在癌细胞系中crispr/cas9筛选发现,另外的肿瘤抑制物改变几乎没有优点,并且可能甚至是有害的。这一发现与处于更成熟的进化状态的癌细胞系一致,由于它们的晚期疾病来源以及针对培养中的最佳增殖能力的选择,接近最佳生长适合度。此外,肿瘤抑制和体内环境的很多方面之间的密切联系强调了分析肿瘤抑制物丧失在体内肿瘤(或例如在组织(诸如类器官培养或3d培养组织)的情形中)中的作用的重要性。有趣的是,在人癌症中肿瘤抑制物改变的频率不直接对应于它们的肿瘤抑制物功能的大小。例如,setd2和rbm10在类似百分比的人肺部腺癌中是突变的,但是setd2缺陷与rbm10缺陷相比,带来更大的生长有益效果(图5a、b)。这突出了对允许快速和定量分析体内基因功能的方法的不断需求,这些分析确定了低频率推定肿瘤抑制物的功能重要性,这些肿瘤抑制物对于单独的患者可以是非常重要的。对肿瘤抑制物改变的临床重要性的理解非常有限,并且这仍然是主要的未满足需求,但是肿瘤生长的强驱动子可以表示比弱驱动子更具吸引力的临床靶标。tuba-seq允许进行对肿瘤抑制基因丧失的更复杂组合的研究,以及肿瘤生长和进展的其他方面的分析。tuba-seq还适用于研究其他癌症类型,并且应允许对通常促进而不是抑制肿瘤生长的基因的研究。最后,该方法允许进行对基因型特异性治疗响应的研究,该反应最终可以产生更精确和个性化的患者治疗。该研究中病灶的统计学性质肿瘤大小的分布通常呈对数正态,当在小鼠-sgrna对内观察时倾向于2阶幂律(图20)。在我们的研究中,给每个肿瘤分配了由具有该肿瘤的小鼠m确定的对数转换大小tmrb、通过第一条形码鉴定的同源sgrnar以及唯一条形码序列(dada2聚类的共有区)b。我们的方法被设计为查询和解决许多来源的误差:我们发现(i)经由相同的插管程序用相同的慢病毒滴度感染(转导)的重复小鼠(通常是同窝仔)中的刺激的肿瘤数大不相同,(ii)重复小鼠中的平均肿瘤大小各不相同,(iii)某些小鼠更适合具有靶向特定肿瘤抑制物的sgrna的肿瘤的生长,并且(iv)在相同的小鼠中具有相同的sgrna的肿瘤的大小大不相同。总体而言,肿瘤抑制物失活对肿瘤负荷的作用比这些其他来源更小。现有的基于病毒-cre的基因工程小鼠模型通过启动成百上千个肿瘤/小鼠解决了差异的主要来源。即使在这种情况下,我们也观察到在相同的小鼠中用相同的遗传构建体刺激的肿瘤大小的随机性。从未准确测量这些实验中单独的肿瘤中的癌细胞数;相反,最常测量的是肿瘤总面积,它是平均肿瘤大小和刺激的肿瘤数的合并。因此,该方法是有缺陷的,因为(i)采样平均大小不是平均大小的最佳估算值,(ii)从未直接测量到刺激的肿瘤数(该数量是变化的,变异系数(cv)为27%),(iii)忽略了小鼠背景的差异,并且(iv)用于评估肿瘤面积的方法也引入了差异。由于这些原因,在krasg12d/+背景(setd2)中,即使最强大的肿瘤抑制物的作用大小也小于重复小鼠之间的方差(图6c)。在单独的小鼠中用病毒-cre载体刺激的病灶数的差异也会影响肿瘤抑制物的作用的估算值。通过对每个肿瘤进行唯一条形码编制,然后使用我们在“方法”中详述的计算方法精确识别肿瘤,我们可以使该差异来源最小化。在我们的流程中识别的病灶数的差异在重复测序运行中表现出cv为10.7%,而重复小鼠之间的识别的病灶的差异表现出cv为27%。因此,我们的基于唯一dna条形码的肿瘤数估算值与重复小鼠之间的肿瘤数是常数的假设相比显著更精确(由于感染(转导)的技术差异,可能具有不同的被慢病毒载体感染(转导)的上皮细胞数)。下面,我们查询、减少和讨论了上表中列出的其余差异来源。与sgrna无关的小鼠间差异我们的多重方法查询了可归因于以下各项的生长作用:(i)crispr/cas9-靶标肿瘤抑制基因,(ii)单独的小鼠,和(iii)它们的相互作用。这仅仅是可能的,因为我们包括了每只小鼠中的很多sgrna,并且测量了相同的小鼠中具有相同的sgrna的很多病灶。我们观察到每个sgrna的平均、对数转换、偏差校正的预期大小中,表面上重复小鼠之间的统计学上显著的差异(ηmr=emr[tmrb])。可以简要汇总这些差异,然后从tmrb中减去,以更好地解析每个肿瘤抑制物的强度。小鼠表现出与sgrna无关的生长干扰ηm=er[ηmr](即,存在一系列肿瘤易感性和肿瘤抵抗性小鼠)以及小鼠中的sgrna依赖性协方差ηmr(具有异常大的lkb1缺陷肿瘤的小鼠也具有异常大的setd2缺陷肿瘤)。通过校正归一化ηm消除了约40%的小鼠间差异,而预计不归因于与sgrna无关的因素的ηmr差异仅为10.7%。我们可以仅消除五分之一的这些ηmr差异(详情如下)。因此,大多数肿瘤易感性差异似乎与sgrna无关的,然而当将平均肿瘤生长优势估算至精密度<10%时,仍然会产生微小的基因-小鼠协方差的结果。重复小鼠,即在肿瘤启动后的相同时间点分析的具有相同的基因工程元件的那些小鼠,通常是同窝仔和同笼仔,但是来源于混合129/bl6背景。虽然这些小鼠可能具有比现实患者更同质的基因型和环境,但是单独的小鼠之间的相关差异仍然存在。重要的是需要注意,虽然由于我们的前所未有的分辨率,在我们的数据中可以鉴定出这些趋势,但是差异很小并且对比较不同的小鼠构建体的实验(例如比较具有和不具有所关注的基因的floxed等位基因的小鼠中的肿瘤生长的常规方法,或我们自己的来自具有lenti-sgsetd2/cre与lenti-sgneo/cre的肿瘤的小鼠的结果(参见图6))应具有甚至更大的作用。因为每只小鼠含有若干无活性sgrna(它们的平均值在小鼠中彼此无明显差异),我们仅通过使相对于聚集的sgrna平均值的大小归一化即可扣除与sgrna无关的小鼠的作用:μmr=emr[tmrb]–em,inerts[tmrb]。在我们的非参数方法中,我们只需要除以中位数无活性sgrna即可,这往往与lnmle平均值几乎相同。sgrna特异性小鼠间差异在单只小鼠中多个活性sgrna的可用性允许我们查询sgrna特异性小鼠的作用。总体而言,μmr基质在kt;cas9小鼠的活性sgrna之间的是高度正相关的。我们使用主成分分析(pca)分解了这些相关性。第一主成分(pc1)解释了重复kt;cas9小鼠中75%的μmr差异。我们测试了对于该协方差的若干假设:1.具有平均较大的肿瘤的小鼠还可能具有对数尺度上的较大的肿瘤差异。如果是这样,则sglkb1与sginert肿瘤大小的比率将与sgsetd2与sginert肿瘤大小的比率共变化。2.小鼠性别驱动了这些不同的生长模式。3.cas9核酸内切酶切割效率在h11lsl-cas9/+小鼠与h11lsl-cas9/lsl-cas9小鼠之间是不同的。4.小鼠中的未知遗传或环境因素会干扰驱动子子集的强度。我们通过将pc1与平均肿瘤大小进行比较以及通过将我们的kt;cas912周队列中的kt;h11lsl-cas9/+与kt;h11lsl-cas9/lsl-cas9进行比较来研究了这些假设中的前两个。pc1与平均肿瘤大小(经由我们的流程所计算)和肺部重量二者具有很好的相关性(图20b-d)。肺部重量(单位为克)在肺部样品收集时测定,并且可能受到肿瘤数量和平均肿瘤大小的影响。肺部重量与pc1(如平均肿瘤大小)的相关性确保了这些观察的趋势不是流程假象。小鼠性别还与pc1(点二系列相关性r=0.75,数据未示出)共变化,并且与我们的第一假设一致,因为雄性小鼠表现出较大的肿瘤以及强驱动子和inert之间较大的大小差异。在12周kt;cas9小鼠中,h11lsl-cas9等位基因状态(杂合或纯合)与pc1(r=0.34,数据未示出)无统计学上显著相关性。因此,我们不认为对于h11lsl-cas9等位基因是杂合的还是纯合的在实质上有助于基因失活的功效。最后,潜在遗传或环境因素的假设过于开放,以至于无法在此处进行检验。然而,我们的方法允许推动对这些因素的研究。因此,我们得出结论,肿瘤容许性和小鼠性别是重复小鼠之间的这些sgrna特异性差异的主要原因,并且cas9核酸内切酶杂合性似乎不会显著影响肿瘤生长,我们的分析流程的结果与其他小鼠测量值一致。概率主成分混合模型被用于从μmr中消除ηmr。该模型确定了从与重复队列中的其他项相同的分布所产生的小鼠的对数似然。本质上,该模型鉴定了具有异常sgrna谱的小鼠。然而,我们只是根据异常的可能性对每只小鼠进行加权,而不是将小鼠归类为‘异常’或‘可接受’的小鼠。在统计学上,‘异常’被定义为似乎从与队列不同的分布得出的点。实际上,我们发现使用马氏(mahalanobis)距离—用于鉴定多维数据中的异常项的常用指标鉴定到类似的异常小鼠。然而,马氏距离指标需要用于对在我们的应用中是特殊的(adhoc)异常项进行分类的一些阈值。使用我们的概率主成分混合模型对小鼠进行加权使kt;cas9小鼠的er[μmr]差异减少2.1%。虽然这只是轻微的改善,但是我们将在sgrna赋予的平均生长优点的最终报告中包括该校正,因为我们认为该值应说明所鉴定的差异的所有来源。在重复小鼠的队列中每个sgrna的最终报告的平均生长作用是在我们的混合模型p(m;μmr)中每只小鼠m的似然加权的所有小鼠中的μmr的算术平均值,即我们的参数和非参数方法以及统计学测试全面测量肿瘤生长的大小谱,然后鉴定所述谱背后的外源性因素提供了如下难题:生长优点可以通过解决每个已知的定量问题的肿瘤大小的高度加工的度量来汇总,或者生长优点可以以作出更少假设的方式更明确地汇总。我们选择了两个极端。在任一情况下,定性结论几乎没有太大的差异;然而,我们提供了两种方法,因为该信度是令人鼓舞的,并且因为不同的方法可能会吸引不同的敏感性的读者。我们的基于最大似然估计的方法在以上部分详细描述。总之,它试图解释我们对大小分布的对数正态形状,以及(i)刺激的肿瘤数、(ii)总体肿瘤容许性和(iii)sgrna特异性差异的小鼠间差异的理解。它利用了我们的大小测量的多维性,并且校正了我们发现的每个已知的外源性因素。下面,我们讨论了假设对数正态的局限性,并且将参数方法延伸至表现出幂律尾部的肿瘤抑制物分布。我们的非参数汇总提供了t(非参数)mrb的百分位(上文定义),以测定大小分布中各种位置处的肿瘤生长增加。它不假设肿瘤分布的形状,并且不对小鼠间差异建模。但是通过校正inert的中位数大小和每只小鼠中存在的肿瘤数,消除了大多数小鼠间差异。由于这个原因,在第一个实验之后,总是报告相对于其对应的inert百分位的百分位。预测和观察针对sgrna的不同百分位等级之间的自相关;不同的百分位等级不是统计上独立的值,并且我们未部署假设它们的独立性的统计学测试。非参数方法通常会发现,第90个至第99个百分位的活性sgrna分布与inert具有最大偏差。我们的发现,即分布至少是对数正态偏态的,与该现象一致。此外,活性sgrna可以引入应模拟inert分布的框内插入和缺失,所以我们预期活性sgrna分布中的最小肿瘤—具有框内突变或无突变—可模拟inert大小。最后,单个无效等位基因的单倍剂量不足通常是未知的,但是如果单倍剂量不足是部分显性或不存在的,则大小分布将在较高的(第90个至第99个)百分位产生最大偏差。所以,我们使用第95个百分位作为驱动子的生长有益效果的粗略汇总,因为它大致平衡了我们的无效突变率、接合性、统计学分辨率(它在较高百分位处会降低)的问题,以及我们对总体大小分布的理解。我们的数据表明,肿瘤抑制物的丧失并不一定在所有单独的肿瘤中产生生长优点(例如图1和2中的p53缺陷与lkb1缺陷)。实际上,出于与p53丧失和肥尾分布的预期结果一致的原因,第95个百分位度量不能在我们的实验中检测p53。然而,简化可以是有用的,并且第95个百分位的大小很好地汇总了生长差异。所有置信区间和p值都经由tmrb的自举来获得。在自举采样之后,针对每次自举(归一化为无活性、pca等)重新计算我们的分析流程中的所有后续步骤。自举样品的大小等于每个实验的原始tmrb(例如,在肿瘤启动后12周分析kt;cas9小鼠中的肿瘤)并且进行重置采样。针对报告的每个95%置信区间采集200000个样品,并且针对报告的每个p值采集2000000个样品。比率的置信区间反映了活性sgrna分布和无活性sgrna分布二者中的不确定性。因此,当sgrna比率的置信区间不包含1时,可以拒绝sgrna的该汇总统计与无活性sgrna匹配的零假设,p<0.05(假设没有针对多假设的校正)。所有p值都报告了以下双侧假设:sgrna汇总统计不同于无活性sgrna汇总统计,并且对于我们的以下多假设是邦费罗尼校正的:11个活性sgrna中的任一个都可以产生生长优点或缺点。虽然活性sgrna始终相当于整体sgrna分布(四个不同的无活性sgrna),但是无活性sgrna仅相当于其他三个inert的分布。大于0.0001的p值未报告,因为当限于2000000个样品时,这是自举的分辨率极限。大小分布的综合参数描述病灶大小大致呈对数正态分布,并且一些基因型中存在过量的非常大的病灶。对于每种基因型和时间,我们将多种2-3个参数的概率分布拟合到观察的病灶大小分布:(log)-正态、(log)-γ、(log)-逻辑、指数、β、广义-极值(包括耿贝尔(gumbel))和幂律(包括帕累托(pareto))。所有病灶大小分布与对数正态、log-γ或log-逻辑分布中的任一者最佳拟合,但是没有一个单分布优于其他分布。柯尔莫哥洛夫-斯米尔诺夫(kolmogorov-smirnov)检验通常会拒绝最佳拟合的单分布—即在很多情况下可以仅存在最小不当拟合。这种缺点突出了我们能够首次测量的大量肿瘤大小以及肿瘤进展的复杂性。因此,我们研究了多族参数拟合。对于一些分布,我们的数据最好地描述了对数正态和幂律尺度变换的组合。虽然log-γ和log-逻辑拟合有时优于对数正态拟合,但是这些替代的分布仅具有更快增长的高阶矩,这暗示了幂律行为。此外,log-γ和log-逻辑分布参数的最大似然估算值必须在数值上求解,而不能保证收敛性。谨慎无偏向地鉴定幂律分布。使用最大似然对潜在的幂律拟合进行参数化,并使用边缘似然对其进行裁定:1.鉴定了对于正实数的整体支持的每个sgrna分布的最大似然对数正态拟合这里,表示对数正态分布的概率密度。2.鉴定了对于在定义域[x(min)r,∞)中的肿瘤的最大似然幂律拟合:这里,表示指数为αr的幂律或帕累托分布的概率密度,并且使用步骤1的对数正态拟合。请注意,当x(min)=0时,幂律未定义,所以习惯在自由浮动最小值的有限支持下检验幂律。3.使用边缘似然来裁定多重拟合模型:使用贝叶斯信息准则(bayesian-informationcriterion,bic)校正模型的自由度的观察数据的似然。该方法是alstot等人推荐的,并且他们随附的软件包被用于进行该分析。最大似然拟合的细节在图3中提供。报告的p值是联合对数正态和幂律拟合的边缘似然的转换,以使得p=1/(1+exp[边缘似然])。这些值检验了以下零假设:数据在整体支持中呈对数正态分布。我们还检验了以下假设:大小根据指数截尾的幂律来分布。该比较是普通幂律的常见反假设,并且意味着无尺度行为仅在有限间隔上存在3。我们未发现针对指数截尾的幂律行为的很好的证据(数据未示出)。由于这个原因,我们认为在以下讨论的基因型中,数据支持肿瘤进展的无尺度模型。我们观察到强大的反复出现的证据,即p53缺陷型肿瘤呈大尺度幂律分布。在krasg12d/+/p53δ基因型的所有化身中观察到幂律动力学(kpt肿瘤和两个kt;cas9sgp53肿瘤时间点)。针对所有这些幂律分布拟合的边缘似然是良好的或极好的。该信度强烈支持以下假设:krasg12d/+/p53δ基因型在肿瘤大小方面呈幂律分布。通常,幂律拟合的ml指数为大约二(α~2)。假设使用幂律动力学来解释癌症发生率,然而未对肿瘤大小分布进行很好的研究,因为测试该假设的测量以前是非常耗时和昂贵的,所以我们研究了简单进化模型,该模型在下一部分中得到大小的幂律分布。我们的病灶大小的深度查询被证实不仅在精确鉴定驱动子生长优点方面是有用的,而且在发现它们的潜在作用模式的各方面也是有用的。另外的很少获得的驱动子突变预测了肿瘤大小的幂律分布p53缺陷型肿瘤表现出在其最右侧尾部的大小呈幂律分布(图3d)。幂律分布通常不由单步马尔可夫过程产生,相反,由复合随机过程,例如游走或吸积过程产生6。对于这种观察到的幂律分布的最简单的(也是我们认为最有可能的)解释是指数过程的组合,即在指数扩增的p53缺陷型肿瘤中第二驱动子事件的罕见获得。假设肿瘤大小n(t)最初随时间t的推移以速率r1指数生长,以使得令n(t=0)=1,即在感染(转导)时(定义为t=0)有一个肿瘤发生细胞。此外,我们假设在时间t*时,在肿瘤群体中出现具有新驱动子的新克隆,并且以更快的速率r2生长,以使得该克隆在处死时tf在肿瘤群体中占优势地位,即r2(tf-t*)>>r1tf。请注意0≤t*<tf。最后,假设该转化克隆及时随机出现,概率与肿瘤的大小成比例,即p(t*)~μn(t)。在这种情形下,分析时的肿瘤大小n(t=tf)=n为根据newman,m.powerlaws,paretodistributionsandzipf’slaw.contemp.phys.46,323–351(2005),标题为combinationsofexponentials(第4.1节)综述的推导过程,我们发现:肿瘤大小呈幂律分布,指数为该结果暗示以下结论中的任一者:观察到的指数必须小于2,或必须获取另外的驱动子。针对在11周处死的kpt小鼠,指数的最大似然估算值略大于二,而在15周处死的kt;cas9小鼠中针对sgp53肿瘤的指数略小于二(虽然这二个值在95%ci中包含二)。假设描述n(t)=ert指数生长动力学p(t*)~μn(t)第2驱动子产生,其中概率与群体大小成比例r2(tf-t*)>>r1tf第2驱动子在处死时完成选择性清除所有上述假设均在肿瘤进展的其他基本数学模型中作出4。因此,我们认为马尔可夫过程是观察到的幂律的最佳解释。最后,我们注意到在时间t*时的转化事件未指定。它可以是遗传改变、表观遗传变化、细胞信号传导状态转换等。我们还注意到,还存在可以产生幂律分布的其他过程。多次慢病毒感染的肿瘤的证据大小测量足以精确鉴定推定由多次慢病毒载体感染(转导)的病灶。我们的第一个实验(kt、klt、kpt小鼠)使用较大的病毒滴度(6000至22000个衣壳),所以我们预期多次感染更为普遍。如果两个不同的病毒载体被相同的创始细胞感染(转导),则它可扩增为以两个病灶—通过两个慢病毒条形码注释的单个肿瘤。所以,如果我们在单独的小鼠中观察到相同大小的两个具有条形码的肿瘤,则我们可以预期这些肿瘤由启动单个病灶的两个慢病毒载体产生。因此,我们研究了每个病灶和其在相同小鼠中的最近邻近者之间的大小差异。我们观察到一小部分病灶的大小比预期更接近,这表明一些病灶可以由用多于一个慢病毒载体的感染(转导)所启动之处的细胞产生。我们的(无效)预期分布表示观察的病灶大小及其在不同(随机选择)小鼠中的最近邻近者之间的大小差异。虽然我们的数据表明,多次感染出现在约1%的肿瘤中,我们不认为这种罕见的存在会严重影响我们研究的其他结论,因为(i)多次感染似乎是罕见的,以及(ii)多次感染应削弱我们的驱动子生长有益效果估计(因为多次感染将赋予我们的基线—sginert构建体以生长优点)。然而,该初步发现再次展示了我们的方法使用旧技术来发现新生物学的能力。方法小鼠和肿瘤启动kraslsl-g12d(k)、lkb1flox(l)、p53flox(p)、r26lsl-tomato(t)和h11lsl-cas9(cas9)小鼠已有所描述。如上文所述,使用指定滴度的慢病毒cre载体通过小鼠的气管内感染(转导)启动肺部肿瘤。通过所示的荧光显微镜检查、肺部重量和组织学来评估肿瘤负荷。根据斯坦福大学实验动物护理和使用委员会(stanforduniversityinstitutionalanimalcareandusecommittee)的指导原则来进行所有实验。具有条形码的lenti-mbc/cre和lenti-sgpool/cre载体库的产生为了能够使用高通量测序来平行定量单独的肿瘤中的癌细胞数,我们使用短条形码序列将慢病毒-cre载体多样化,由于慢病毒载体稳定整合进初始感染(转导)的肺上皮细胞,该短条形码序列是每个肿瘤唯一的。我们使用两种不同的具有条形码的慢病毒载体库在很多小鼠背景下产生肿瘤。第一个库是约106个具有唯一条形码的lenti-pgk-cre变体(lenti-millionbc/cre;通过合并六个具有条形码的lenti-u6-sgrna/pgk-cre载体来产生的lenti-mbc/cre)的库,我们使用该库来分析在kraslsl-g12d/+;r26lsl-tomato(kt)、kraslsl-g12d/+;p53flox/flox;r26lsl-tomato(kpt)和kraslsl-g12d/+;lkb1flox/flox;r26lsl-tomato(klt)小鼠中诱导的肿瘤的癌细胞数(图1)。第二个库是15个具有条形码的lenti-u6-sgrna/pgk-cre载体的库,我们使用该库通过感染kt;h11lsl-cas9(kt;cas9)和kt小鼠来评估候选肿瘤抑制基因在三种不同的遗传背景下的肿瘤抑制作用。我们的lenti-sginert/cre载体包含靶向rosa26lsl-tomato等位基因中的neor基因的三个sgrna,该sgrna是活性切割、但在功能上是无活性的阴性对照sgrna。sgrna的设计、产生和筛选我们产生了携带cre的慢病毒载体以及靶向11个已知和推定的肺部腺癌肿瘤抑制物sglkb1、sgp53、sgapc、sgatm、sgarid1a、sgcdkn2a、sgkeap1、sgrb1、sgrbm10、sgsetd2和sgsmad4中的每个的sgrna。还产生了携带无活性指导的载体:sgneo1、sgneo2、sgneo3、sgnt1和sgnt3。鉴定了所有可能的靶向所关注的每个肿瘤抑制基因的20-bpsgrna(使用nggpam),并使用可用的sgrna设计/评分算法对预测中靶切割效率进行评分10。对于每个肿瘤抑制基因,我们选择预测为最可能产生无效等位基因的三个唯一sgrna;优先选择具有最高预测切割效率的sgrna,以及靶向在所有已知的剪接亚型(ensembl)中保守的、最接近剪接接受体/剪接供体位点、最早定位在基因编码区中、出现在注释功能结构域上游(interpro;uniprot)以及出现在已知的人肺部腺癌突变位点上游的外显子的那些sgrna。如上文所述产生含有每种sgrna的lenti-u6-sgrna/cre载体。简而言之,q5定点诱变(nebe0554s)被用于将sgrna插入含有u6启动子以及pgk-cre的亲本慢病毒载体。通过用每种lenti-sgrna/cre病毒感染lsl-yfp;cas9细胞来确定每种sgrna的切割效率。在感染(转导)后四十八小时,使用yfp阳性细胞的流式细胞定量来确定感染(转导)百分比。然后从所有细胞提取dna,并通过pcr扩增被靶向的肿瘤抑制基因的基因座。对pcr扩增子进行桑格测序,并使用tide分析对其进行分析,以定量插入缺失形成的百分比。最后,如通过流式细胞术所确定,用通过tide确定的插入缺失的百分比除以lsl-yfp;cas9细胞的感染(转导)百分比,以确定sgrna切割效率。将靶向所关注的每个肿瘤抑制基因的最有效的sgrna用于后续实验。靶向tomato和lkb1的sgrna此前已有所描述,并且我们此前验证了靶向p53的sgrna(数据未公开)。用于扩增针对本研究所用的最前端指导的靶标插入缺失区的引物序列如下:f引物(5’→3’)r引物(5’→3’)sgapc_1tgactttgcagggcaagtttcccactcccctgttacctttsgarid1a_3cagcagtccccaactccataggagccatttcttggggttasgatm_3gccccaagtgagaatcagtgagctctggctccttgtggatsgcdkn2a_2ggcttctttcttgggtcctgggctcatttgggttgcttctsgkeap1_2ctgagccagcaactctgtgaggcctatcccacttctgagcsgrb1_3aactgtgctggtgtgtgcaaacaccaccaccaccatcatcsgrbm10_3caaagctggaagcgagactgctggctggagctgtgagagtsgsetd2_1tctgcaagttcaagcgatgatggattcaggtgacctagatggsgsetd2_2cctccagccgctcctcatgaacgccgaacctaagcagsgsmad4_3gcctttctgtggaaatggaattccaggctgagtggtaaggsgneo_1ttgtcaagaccgacctgtccccaccatgatattcggcaagsgneo_2tctggacgaagagcatcagggctccaatccttccattcaasgneo_3cgctgttctcctcttcctcatggatactttctcggcaggalenti-sgrna/cre的条形码多样化在鉴定靶向所关注的每个肿瘤抑制物的最佳sgrna之后,我们用对每个单独的sgrna具有特异性的已知的8个核苷酸的id(sgid;以粗体表示)和15个核苷酸的随机条形码(bc;以下划线表示)使对应的lenti-sgrna/cre载体多样化(参见图10a)。这些引物被用于pcr扩增包含pgk启动子的3’末端和cre的5’部分的lenti-pgk-cre载体的区域。使用hsdna聚合酶(预混合)(clontech,r040a)进行pcr,并且使用pcr纯化试剂盒(28106)来纯化pcr产物。pcr插入物被bspei和bamhi消化,并与lenti-sgrna-cre载体连接,用xmai(产生bspei相容性末端)和bamhi切割该载体。为了产生大量具有唯一条形码的载体,我们使用t4连接酶(neb,m0202l)和标准方案(80μl总反应体积)将300ng每个xmai、bamhi消化的lenti-sgrna-cre载体与180ng每个bspei、bamhi消化的pcr产物连接。使用pcr纯化试剂盒来pcr纯化连接物,以除去残留的盐。为了获得最大可能数量的具有唯一条形码的lenti-sgrna/cre载体库,将1μl纯化的连接物转化至20μlelectromaxdh10b细胞中(thermofisher,18290015)。在0.1cmgenepulser/micropulser样品池(bio-rad,165-2089)中在bdmicropulsertm电穿孔仪(bio-rad,165-2100)中1.9kv下对细胞进行电穿孔。然后通过添加500μl培养基并在37℃下以200rpm振荡30分钟来拯救细胞。对于每次连接,将细菌接种在七个lb-amp平板(1个平板1μl,1个平板10μl,并且5个平板100μl)上。第二天,在1μl或10μl平板上对菌落进行计数,以估算100μl平板上的菌落数,该菌落数可用作与每个id相关的唯一条形码的初始估算数。将10ml液体lb-amp添加至每个细菌的平板以合并菌落。将菌落从平板上刮入液体中,将来自每次转化的所有平板组合到烧瓶中。将烧瓶在37℃下以200rpm振荡30分钟以混合。使用hispeedmidiprep试剂盒(12643)来midi制备dna。使用qubitdsdnahs试剂盒(invitrogen,q32851)来测定dna浓度。作为质量对照度量,用gotaqgreen聚合酶(promegam7123)按照制造商的说明来pcr扩增来自每个lenti-sgrna-sgid-bc/cre质粒库的sgid-bc区。对这些pcr产物进行桑格测序(stanfordpan设施)以确认预期的sgid和随机bc的存在。由于bspei和xmai具有相容的突出端,但具有不同的识别位点,因此从sgid/bc的成功连接产生的lenti-sgrna-sgid-bc/cre载体缺乏xmai位点。因此,对于如通过桑格测序所测定具有可检测量的无条形码的亲本lenti-sgrna/cre质粒的库(>5%),我们通过使用标准方法消化具有xmai的库(neb,100μl反应),从而破坏了亲本无条形码的载体。使用pcr纯化试剂盒来再纯化这些再消化的质粒库,并且通过nanodrop来再确定浓度。lenti-mbc/cre和lenti-ts-pool/cre的产生为了获得具有大约106个相关条形码的文库(该文库可用于我们的初始实验的缺乏h11lsl-cas9等位基因的小鼠中),我们合并了六个sgid-bc具有条形码的载体,从而产生lenti-millionbarcode/cre(lenti-mbc/cre)。然后我们合并具有条形码的lenti-sgrna-sgid-bc/cre载体(sglkb1、sgp53、sgapc、sgatm、sgarid1a、sgcdkn2a、sgkeap1、sgneo1、sgneo2、sgneo3、sgnt1、sgrb1、sgrbm10、sgsetd2和sgsmad4),从而产生lenti-sgts-pool/cre。在慢病毒产生之前如通过qubit浓度所确定以等比率合并所有质粒。慢病毒的产生、纯化和滴定使用通过慢病毒载体和delta8.2和vsv-g包装质粒进行的基于聚乙烯亚胺(pei)的293t细胞转染来产生慢病毒载体。针对肿瘤启动而产生lenti-mbc/cre、lenti-sgts-pool/cre、lenti-sgtomato/cre、lenti-sglkb1、lenti-sgsetd2#1/cre、lenti-sgsetd2#3/cre、lenti-sgneo2/cre和lenti-sgsmad4/cre。在转染后八小时,以0.2mm的终浓度添加丁酸钠(sigmaaldrich,b5887),以增加病毒粒子的产生。在转染后36、48和60小时收集含病毒培养基,通过超速离心(25000rpm1.5-2小时)来浓缩,重悬于pbs中过夜,并冷冻于-80℃下。通过感染lsl-yfp细胞(alejandrosweet-cordero博士馈赠),由流式细胞术测定yfp阳性细胞的百分比,以及将感染滴度与已知滴度的慢病毒制备物进行比较来对浓慢病毒粒子进行滴定。“基准”细胞系的产生使用三个具有sgid“ttctgcct”的具有唯一条形码的lenti-cre载体来产生可以以已知的细胞数加标至每个成批肺部样品的基准细胞系,从而能够计算每个肿瘤中的癌细胞数。使用qiaprepspinminiprep试剂盒(27106)来分离来自单独的细菌菌落的质粒dna。对克隆进行桑格测序,如上文所述的产生慢病毒,并且以非常低的感染(转导)复数来感染(转导)lsl-yfp细胞,以使得在48小时后大约3%的细胞是yfp阳性的。扩增感染(转导)的细胞,并使用bdariaiitm(bdbiosciences)对所述细胞进行分选。接种并扩增yfp阳性分选的细胞,以获得大量细胞。在扩增后,在bdlsriitm分析仪(bdbiosciences)上再次对细胞进行yfp阳性细胞的百分比分析。使用该百分比,针对三种细胞系中的每种计算包含5x105个整合的具有条形码的慢病毒载体所需的总细胞数,并且根据该计算将细胞分成等份并冷冻。所有小鼠感染的汇总基因型病毒类型病毒滴度ktlenti-mbc/cre6.8x105ktlowlenti-mbc/cre1.7x105kptlenti-mbc/cre1.7x105kltlenti-mbc/cre1.7x104ktlenti-ts-pool/cre9.0x104kt;cas9lenti-ts-pool/cre2.2x104kt;cas9lenti-sgneo2/cre9x103kt;cas9lenti-sgsetd2#1/cre9x103kt;cas9lenti-sgsetd2#2/cre9x103ktlenti-sgsmad4/cre105kt;cas9lenti-sgsmad4/cre105来自小鼠肺部的基因组dna的分离对于其中将条形码测序用于定量每个肿瘤中的癌细胞数的实验,使用fishertissuemeiser来对每只小鼠的全肺进行匀浆。在匀浆时添加来自三种单独的具有条形码的基准细胞系中的每种的5x105个细胞。在20ml裂解缓冲液(100mmnacl、20mmtris、10mmedta、0.5%sds)中用200μl20mg/ml蛋白酶k(lifetechnologies,am2544)来对组织进行匀浆。将匀浆的组织在55℃下温育过夜。为了维持所有肿瘤的准确呈现,使用标准方案用酚-氯仿提取dna,并用乙醇从约1/10的总肺裂解物沉淀dna。对于重量小于0.3克的肺,从约1/5的总肺裂解物提取dna,对于重量小于0.2克的那些肺,从约3/10的总肺裂解物提取dna,以增加dna收率。用于测序的sgid-bc文库的制备物通过扩增每只小鼠32μg基因组dna的sgid-bc区来制备文库。使用24个引物对之一来pcr扩增整合lenti-sgrna-bc/cre载体的sgid-bc区,所述引物对含有truseq衔接子和5’多重标签(truseqi7索引区以粗体表示):我们使用sgid-bc区的单步pcr扩增,我们发现所述pcr扩增是测定每个肿瘤中的癌细胞数的高度可重现的定量方法。我们使用onetaq2xmaster混合物与标准缓冲液(neb,m0482l)通过以下pcr程序来进行八个100μlpcr反应/小鼠(4μgdna/反应):1.94c10min2.94c30s3.55c30s4.68c30s5.转到2(34x)6.68c7min7.4c无穷大通过凝胶电泳来分离合并pcr产物,并使用minelute凝胶提取试剂盒来提取凝胶。通过生物分析仪(agilenttechnologies)来测定来自单独的小鼠的纯化pcr产物的浓度并以等比率合并。在hiseq上对样品进行测序,以产生100bp的单端读段(elimbiopharmaceuticals,inc)。经由超深度测序来鉴定唯一sgrna和肿瘤唯一sgid-bc鉴定肿瘤。在hiseq上经由下一代测序来检测这些sgid-bc。就细胞数而言,预期每个肿瘤的大小大致对应于每个唯一的sgid-bc对的丰度。因为肿瘤大小的变化系数大于读段测序误差率,所以从反复出现的读段误差区分出真实肿瘤需要对深度测序数据的仔细分析。通过三个步骤来鉴定肿瘤及其各自的sgrna:(i)丢弃来自超深度测序运行的异常和低质量读段,(ii)将唯一条形码堆积捆扎成我们预测来自相同的肿瘤的组,以及(iii)以被证实为最可重现的方式从这些捆束估算细胞数。读段预处理读段含有双组分dna条形码(8个核苷酸的sgid和含有15个随机核苷酸的21个核苷酸的条形码序列),该条形码从我们的正向引物下游的49个核苷酸开始,至我们的100-bp单端读段的末端上游的22个核苷酸结束。我们丢弃了不寻常的读段:缺乏侧接的慢病毒序列的那些读段、含有意外的条形码的那些读段以及具有高误差率的那些读段。这通过三个步骤实现(图8a):1.我们检查了紧邻sgid-bc的上游和下游的12个慢病毒核苷酸。使用邻近的6聚体搜索串对来鉴定这12个核苷酸,以使得每个6聚体可以容忍一个错配。虽然我们预期这12个核苷酸从读段中的第37位开始,但是我们不要求这种定位或不利用此信息。巢式6聚体方法(具有两次机会来鉴定侧接sgid-bc的慢病毒序列)可用于最大程度地减少读段丢弃。这在第一步中是特别重要的,因为我们的读段的非条形码区被用于估算测序误差率,所以不应针对读段误差而偏差。对于约7-8%的读段,这种第2个6聚体匹配挽救了读段,即紧密侧接sgid-bc的6聚体不如预期(尽管我们容忍一个错配),但是紧邻这些内部6聚体序列外部的6聚体是可识别的,并允许我们挽救读段和鉴定条形码。2.然后我们丢弃了其中sgid-bc长度在任一方向上偏离大于两个核苷酸的读段。因为预期我们的第一条形码含有15个sgid中的一个,所以我们丢弃了与这15个序列中的一个不匹配的读段。该匹配中允许一个错配和一个插入缺失。3.然后我们对每个读段进行末端修剪,以使得18bp侧接sgid-bc的任一末端。然后我们根据质量得分对修剪的读段进行过滤,保留预测为含有不超过两个测序误差的那些读段。我们还丢弃了在第二(随机)条形码中具有未识别的碱基的读段并纠正了在其他位置未识别的碱基。在这三个阶段中,在第一阶段丢弃14%的读段,在第二阶段丢弃约7%,在第三阶段丢弃<2%。然后我们检查了在每个阶段被丢弃的那些读段。通过执行blast搜索,我们确定在第一阶段丢弃的那些读段通常含有对应于来自我们的制备物(phix噬菌体基因组和小鼠基因组)或在泳道上与我们配对的其他样品(常见质粒dna)的人工产物的非信息序列。在第二阶段,我们发现具有异常条形码长度的读段通常含有大插入缺失,或它们的sgid-bc中的一者或二者完全缺失。最后,在第三阶段,由于以下事实很少丢弃读段:读段的内部区域表现出比对应的末端更高的得分。由于这种趋势,通常在丢弃预计含有多于两个测序误差的那些读段之前对读段实施末端修剪。经由dada2进行唯一读段堆积的聚类将sgid-bc读段聚集成相同序列的组,并进行计数。唯一dna条形码对的计数不直接对应于唯一的肿瘤,因为预期大肿瘤会产生反复出现的测序误差(图8b)。因此,我们花费了大量精力来开发从来自大肿瘤的反复出现的测序误差中区分出小肿瘤的方法(例如,考虑到如果机器具有0.1%-1%的误差率,则1000万个细胞的肿瘤会产生模拟1万-10万个细胞的肿瘤的测序误差堆积)。dada2此前被用于解决涉及超深度测序的条形码编制实验中的此问题。然而,因为它被设计为进行全长illumina扩增子的超深度测序,所以我们必须针对我们的目的对其进行定制和校准。在dada2中,较大堆积的反复出现的测序误差产生的条形码堆积的可能性取决于:1.较大堆积的丰度,2.较小堆积和较大堆积之间的特定核苷酸差异,以及3.在不同位置处较小堆积的平均质量得分。首先试探性考虑(使计算速度最大化)然后经由尼德曼-翁施(needleman-wunsch)算法更精确地考虑(在需要时)因素一和因素二。当测序误差产生较小堆积的概率小于ω时,dada2将聚类分为两个。所以,该值表示用于分裂更大的聚类的阈值。当该阈值较大时,读段堆积会被随意分裂(很多称为肿瘤,可能区分大肿瘤),并且当ω较小时,读段堆积会被有限制地分裂(很少称为肿瘤,可能聚集独特的小肿瘤)。从我们的超深度测序数据可以推断出测序误差的可能性。phred质量得分提供了测序误差率的理论估算,然而这些估算往往在机器与机器间有所不同,并且不考虑我们方案的细节(包括例如经由pcr扩增引入的偶然误差,即使我们使用高保真聚合酶)。通常,dada2将与唯一dna聚类同时估算测序误差率;然而,我们的慢病毒构建体在我们的sgid-bc区的外部具有非简并区,所述sgid-bc区可用于直接估算测序误差率。此外,联合估算误差率和条形码聚类的计算量更大,对我们的整个数据集进行聚类以及对相关的聚类参数进行研究需要超过20000个cpu-小时。针对每个机器训练测序误差模型的步骤为:1.通过将紧邻我们的sgid-bc上游的18个核苷酸与紧邻条形码下游的18个核苷酸连接在一起来产生训练伪读段,然后2.使用dada2的单次运行对这些伪读段进行聚类。3.使用从该训练运行估算的误差率,以便使用dada2的单次运行来对sgid-bc进行聚类。我们使用非常低的值ω=10-100来估算训练运行中的测序误差,因为我们预期只有一个慢病毒sgid-bc侧接序列的聚类。改变该值不会明显影响我们的训练运行,但是尽管如此,我们仍然在该值下从我们的慢病毒序列中偶尔观察到非常小的导数聚类。这些导数聚类大概是罕见的dna人工产物,并且总数从未超过我们处理的读段的2%。我们认为使用非常严格的dada2运行来估算测序误差代表更好的方法(根据goldilocks原则):更大的允许阈值可能会过拟合测序误差并低估测序误差率,而其中从每个读段与预期值的偏差直接估算误差率的方法(类似于其中ω=0的dada2运行)将忽略dna人工产物在我们的数据中的存在,所以高估了测序误差率。我们在该研究中使用的每个机器(总共七个)上训练测序误差率。训练允许估算每种置换类型(a→c、a→t等)的概率。使用可用数据的loess回归来确定作为phred质量得分的函数的误差率(图8c)。通常,误差率比针对颠换的phred质量得分的预测值大大约二至三倍(大约与针对转换的预期值一致)。这种误差率升高是典型的,并且可以反映机器的错误校准和/或pcr期间引入的突变。然后我们使用dada2对通过我们的预处理过滤器的双条形码进行聚类。将非简并慢病毒侧接区的七个核苷酸给予条形码,从而鉴定条形码中的任何插入缺失(在不存在足够的侧接序列的情况下,dna比对算法有时会将插入缺失错误识别为多个点突变)。在聚类期间,我们还要求(i)聚类彼此偏离至少两个碱基(即,min_hamming_distance=2),(ii)新聚类仅在堆积尺寸超出误差处理下的预期值的系数为至少二时形成(min_fold=2),以及(iii)尼德曼-翁施算法只考虑具有至多四个净插入或缺失的比对(band_size=4、vectorized_alignment=false)。这些选择均未对结果产生明显影响,但是它们提高了计算性能,并且提供了条形码聚集到合理大小的肿瘤中的另外的验证。流程的检查和校准我们对第一pcr扩增的、多重dna文库(来自kt、klt和kpt肿瘤)进行一式三份测序,以检查和设计我们的肿瘤识别方法。通过三种方式来查询再现性:(i)针对所有条形码和所有小鼠的估算细胞丰度之间的相关性,(ii)在我们的第一实验中在每只小鼠中每个sgid识别的病灶数的差异,以及(iii)每个sgid的平均大小差异—它在不表达cas9的小鼠中应为常数。因为我们的一式三份运行的读段深度自然变化(预处理后的40.1x106、22.2x106和34.9x106个读段),这三个运行在不同的机器上以不同的测序误差率进行,并且因为我们的初始慢病毒库含有具有不同水平的条形码多样性的六个不同的sgid,所以我们的检查过程中的技术差异非常近似于后续实验的技术差异。在我们的肿瘤大小分析流程中,我们发现:1.在重复运行之间,我们的三个“基准”dna条形码的平均丰度比中位数丰度更具可重现性。因此,该基准读段丰度的平均值(对应于500000个细胞)被用于将读段丰度转换为每个肿瘤中癌细胞的绝对细胞数(图9)。2.忽略来自聚类的共有条形码的具有≥2个误差的读段改善了再现性。通常,条形码聚类中约80%-90%的读段与共有条形码完全匹配,而约5%的读段是来自该读段的单独误差,并且约5%-15%的读段在≥2个误差处偏离。这些具有≥2个误差的读段在重复运行之间的相关性较差,并且妨碍了我们可重复地估算绝对细胞数/肿瘤大小的能力。据推测,这些读段既没有足够的证据被认为是它们自己的病灶,也没有充分证据被算作更大的聚类。因此,这些读段被排除在外。3.dada2的聚类分裂倾向的阈值被定为ω=10-10,并且对于图1-图3,要求病灶含有≥500个细胞,对于图4-图6,要求病灶含有≥1000个细胞,以使重复运行之间的再现性最大化(图8d-f)。具有高特异性的阈值参数(ω越小,最小细胞数越大)以更大的可重复性识别病灶大小,而具有高灵敏度的阈值参数(ω越大,最小细胞数越小)以更大的可重复性识别病灶数量。仅将再现性的一个方面放在过于优先的位置是不明智的。通过两个阈值,考虑测量误差的不同方面,我们可以更好地平衡这些竞争性优先权。通过此流程,我们以若干方式在我们的筛选中查询了条形码的多样性。首先,我们确认该条形码中的核苷酸在a、t、c和g中均匀分布(图10b)。其次,我们未发现重复串(例如,序列aaaaa)过多的证据。再次,我们计算了在我们的慢病毒库中与每个sgid配对的随机条形码数。由于通过我们的条形码连接方法,我们产生了大量每个载体的具有唯一条形码的变体,(参见lenti-sgrna/cre的条形码多样化)我们的慢病毒库中存在的大多数条形码都从未在任何实验中的病灶中检测到(因为多样性远高于总病灶数)。尽管如此,我们仍然从所观察的条形码推断出条形码多样性的量。为了进行这种推断,我们假设在i小鼠中观察到条形码的概率呈泊松(poisson)分布:p(k=i;λ)=λke-λ/k!,其中λr=lr/dr,是我们的整个数据集(已知数量)中针对每个sgidr的识别病灶数lr除以针对每个sgid的唯一条形码的总数dr的比率。通过注意到λr/(1–e-λr)=μ非零,其中μ非零=σi=1∞p(k=i;λr)仅仅是出现一次或多次的每个条形码的出现平均数,我们可以计算dr。在我们的整个数据集中,在相同的小鼠中启动两个不同肿瘤的相同的条形码的平均概率为0.91%。在lenti-mbc/cre实验中,还通过六个sgid来展示良好的条形码多样性。如果条形码多样性低并且条形码通常在小鼠中重叠,则多样性较低的sgid的平均大小将增加—因为具有相同的条形码的两种不同的肿瘤将捆扎在一起。然而,每个sgid的平均大小在重复小鼠中的变化<1%,因此反驳了这种可能性。尽管存在测序误差,我们还通过以两种方式进行深度测序运行来评估我们准确识别sgid的能力:通过在基于原始读段序列的聚类之前鉴定每个读段的同源sgid,或通过在基于聚类的共有序列的聚类之后鉴定同源sgid。使用任一方法,99.8%的读段与相同的同源sgid配对,从而确保可以准确鉴定sgid。我们选择采用后一种方法来进行我们的最后分析。通过充分开发和检查我们的肿瘤识别流程,我们挽救另外十个大小分辨率。我们的三个dna基准(在dna制备最开始时添加至肺部样品)(图9)提供了该分辨率的概览。通过dna基准的唯一sgid和已知的第二条形码可以容易地鉴定dna基准的测序误差。虽然通常会丢弃这些测序误差,但是我们可以将它们处理为普通读段堆积,并且观察到潜在测序误差的性质。如果没有我们的校准分析流程,测序误差就会以约103个细胞的病灶出现;通过我们的流程,这些测序误差以约102个细胞的病灶出现—小于我们的最小细胞阈值(图2a)。更重要的是,我们的流程对技术干扰是稳健的。我们更深入地分析了来自第一实验的两只特定小鼠中两个另外的技术干扰的再现性。首先,以高深度对klt11周小鼠(jb1349)进行测序,然后随机下采样十倍至典型的读段深度(该下采样比我们的整个研究中实际检测到的任何读段深度差异都更引人注目)。在该第一干扰中,病灶大小是高度相关的(图2b)。另外,在两个pcr反应中用不同的多重标签来扩增kt11周小鼠(iw1301)(图2c)。虽然再现性总体良好,但是pcr和多重似乎比读段深度更能阻碍再现性。这些小鼠还显示出两个令人鼓舞的再现性趋势:(i)较大的病灶/肿瘤在重复之间最一致,以及(ii)肿瘤病灶大小的总体形状(直方图)在重复之间的相关性比单独的肿瘤更好(例如,对于iw1301中的每个病灶,r=0.89,而对于图8b的60个直方图桶内的肿瘤丰度,r=0.993)。此第二观察结果暗示我们的技术干扰引入了无偏差噪声。另外,所有相关性都比较对数大小;因为较大的肿瘤之间的相关性更好,所以该转化基本上降低了皮尔逊相关系数。使gc扩增偏差对肿瘤大小识别的影响最小化我们在我们的研究中通过具有肿瘤的小鼠m对应的大小tmrb、通过第一条形码鉴定的同源sgrnar以及唯一条形码序列(dada2聚类的共有区)定义了每个肿瘤b。已知我们的数据呈大致对数正态结构(图3d,数据未示出),我们对大小进行了对数转换和归一化,以使得tmrb=ln(tmrb/emr[tmrb])。这里emr[tmrb]=σbtmrb/nmr是给定小鼠m和sgrnar的预期病灶大小,我们将该符号用于表示期望值。这种表示法—其中从下标删除聚集指数—在全文中使用。gc偏差是微小的:emr[tmrb]的变异系数(cv)为5.0%。该边缘分布仍然表现出对组合条形码序列的gc含量的微小的依赖性,该依赖性可以用eb[tmrb]的4阶最小二乘多项式拟合f4(b)最好地描述(调整r2=0.994)。sgid均设计为具有均衡的gc含量,然而第二条形码包含随机序列。虽然产生条形码的多项式过程使c中等水平最为常见,但是观察到gc含量的一些偏差。f4(b)的最大值在中等gc含量时产生,这表明pcr使扩增偏向中等解链温度的模板dna。我们从对数转换值减去该gc偏差的作用:tmrb=ln[tmrb]–f4(b)。这种校正使肿瘤大小平均改变5%。使用lenti-ts-pool/cre病毒来进行体外切割效率计算用lenti-ts-pool/cre病毒感染(转导)表达cas9的细胞系并在48小时后收获。提取gdna并使用上述引物来扩增靶基因座。靶位点处的插入缺失分析为了确认crispr/cas9诱导的插入缺失的体内形成,使用gotaqgreen聚合酶(promegam7123)和引物对从来自成批肺部样品的基因组dnapcr扩增所关注的每个基因的靶向区,得到适用于双末端测序的短扩增子:f引物(5’→3’)r引物(5’→3’)apccatggcataaagcagttactacatctcctgaacggctggatacarid1accagtccaatggatcagatggggtacccatgtccttgttgatmcacccagttgaccctatcttcccgttttcggaagttgacagcdkn2acaacgttcacgtagcagctcaccagcgtgtccaggaagkeap1ggcttattgagttcgcctacagctgctgcacgaggaagtrb1ggtacccgatcatgtcagagaaaggaacacagctcccacacrbm10tactcagccgctttctttgcgaggatttgttccgcatcagsetd2ctgttgtggttgtgccaaagttttcagtttgagaacagcctttsmad4tcgattcaaaccatccaacacttgtggaagccacaggaatlkb1gggcctgtacccatttgagtgtcccttgctgtcctaacap53catcacctcactgcatggaccaggggtctcggtgacagneo1ggcaggatctcctgtcatctagtacgtgctcgctcgatgneo2cggaccgctatcaggacatagagcggcgataccgtaaagneo3gatcggccattgaacaagatcatcagagcagccgattgtpcr产物通过凝胶提取或使用minelute试剂盒直接纯化。使用qubiths测定法按照制造商的说明来测定dna浓度。对于每只小鼠以相等的比例组合全部14种纯化的pcr产物。使用spriworks(beckmancoulter,a88267)和标准方案,通过每只小鼠的单个多重标签将truseq测序衔接子连接至合并pcr产物上。在illuminahiseq上进行测序,以产生单末端150-bp读段(stanfordfunctionalgenomicsfacility)。将自定义python脚本用于分析插入缺失测序数据。对于14个靶向区中的每个,在所述靶向区的任一侧选择8聚体以产生46个碱基对的区域。要求读段含有两个锚,并且不允许产生测序误差。然后测定两个锚之间的每个片段的长度并与预期长度进行比较。根据插入或缺失的碱基对数对插入缺失进行分类。在每个单独的小鼠中针对每个单独的基因座的插入缺失的百分比计算如下:然后计算三个neo基因座中的平均插入缺失的%,每个其他靶基因座处的插入缺失%归一化为该值,以产生相对于neo的插入缺%失,该插入缺失%绘制于图6a中。使用lenti-ts-pool/cre病毒来进行体外切割效率计算用lenti-ts-pool/cre病毒感染(转导)表达cas9的细胞系并在48小时后收获。提取gdna并使用上述引物来扩增靶基因座(参见靶位点处的插入缺失的分析)。首先,合并所有引物,并使用gotaqgreen聚合酶(promegam7123)进行15轮pcr。然后将这些产物用于使用单独的引物对进行的后续扩增,如上文所述。如上文所述制备测序文库。组织学、免疫组织化学和肿瘤分析将样品固定在4%福尔马林中并用石蜡包埋。利用abcvectastain试剂盒(vectorlaboratories),使用针对tomato(rocklandimmunochemicals,600-401-379)、smad4(abcam,ab40759)和sox9(emdmillipore,ab5535)的抗体对4μm切片进行免疫组织化学。切片用dab进行显色,并用苏木精进行复染。使用标准方法进行苏木精和伊红染色。将用lenti-sgtomato/cre感染(转导)的肺部切片针对tomato进行染色,并将肿瘤评分为阳性(>95%tomato阳性癌细胞)、阴性(无tomato阳性癌细胞)或混合(所有其他肿瘤)。对肿瘤进行分类,并从单张切片通过来自4只独立的小鼠的所有肺叶进行计数。用lenti-sgsetd2和lenti-sgneo诱导的肿瘤面积的定量和肿瘤的条形码测序将来自用lenti-sgsetd2#1/cre、lenti-sgsetd2#2/cre或lenti-sgneo2/cre病毒感染(转导)的小鼠的携带肿瘤的肺叶包埋于石蜡中、切片并用苏木精和伊红进行染色。使用imagej来确定肿瘤面积的百分比。通过癌细胞各自的慢病毒条形码的测序和后续分析来评估在用lenti-sgsetd2#1/cre和lenti-sgneo2/cre感染(转导)的kt;cas9小鼠中的单独的肿瘤中的癌细胞数的分布,如上文所述。针对lkb1和cas9的蛋白印迹分析来自具有lenti-sglkb1/cre启动的肿瘤的kt和kt;cas9小鼠的显微解剖的tomato阳性肺部肿瘤的cas9和lkb1蛋白表达。在ripa缓冲液中裂解样品,并与lds上样染料煮沸。将变性样品在4%–12%bis-tris凝胶(nupage)上运行并转移到pvdf膜上。使用针对hsp90(bdtransductionlaboratories,610419)、lkb1(cellsignaling,13031p)、cas9(novusbiologicals,nbp2-36440)的一抗和hrp缀合的抗小鼠(santacruzbiotechnology,sc-2005)和抗兔(santacruzbiotechnology,sc-2004)二抗对膜进行免疫印迹。具有cas9介导的smad4失活的小鼠的存活分析为了确认可归因于smad4的功能性肿瘤抑制的缺乏,用105个lenti-sgsmad4/cre来气管内感染(转导)kt和kt;cas9小鼠。小鼠在显示出难以评估存活的可见体征时处死。实施例2:致癌性变体的体内多重定量分析人癌症的大规模基因组分析已对被认为启动肿瘤发展和维持癌症生长的体细胞点突变进行了编目。然而,确定特定改变的功能重要性仍然是我们对癌症的遗传决定因素的理解的主要瓶颈。在这里,我们提供了将多重aav/cas9介导的同源定向修复(hdr)与dna条形码编制和高通量测序整合的平台,该平台能够同时研究小鼠中的从头癌症中的多种基因组改变。使用该方法,我们将非同义突变的具有条形码的文库引入成体体细胞中的kras的热点密码子12和13中,以在肺、胰腺和肌肉中启动肿瘤。来自成批肺和胰腺的具有条形码的krashdr等位基因的高通量测序在kras变体致癌性方面发现了令人惊讶的多样性。同时研究精确基因组改变的体内功能的快速、成本效益好的定量方法将会发现对致癌作用的新颖生物学和临床可行性见解。结果为了以定量和相对高通量方式分析多种点突变的体内致癌功能,我们开发了在若干癌症类型的自身小鼠模型中纳入dna条形码编制和高通量测序的针对体细胞aav/cas9介导的hdr的平台(图23a-d)。我们设计、产生和验证了以多重方式将所有可能的kras密码子12和13单核苷酸非同义点突变引入小鼠体细胞的aav载体文库(图23e-g和图27)。每个aav含有靶向kras的第二外显子的sgrna、约2kbkrashdr模板和cre-重组酶(aav-krashdr/sgkras/cre;图23e和图27a-c)。krashdr模板含有野生型(wt)kras或kras的密码子12和13中的12个单核苷酸非同义突变中的一个以及侧接kras的第二外显子的基因组序列。每个krashdr模板还含有sgkras靶序列中的沉默突变和相关的原间隔序列邻近基序(pam*),以防止cas9介导的krashdr等位基因的切割。为了通过来自成批组织的dna的高通量测序来平行定量单独的肿瘤,我们用随机八个核苷酸的条形码使每个krashdr模板多样化,该条形码被工程改造至12和13下游的密码子的摇摆位置(图23e和图27b、c)。aav载体还编码cre-重组酶。cre表达能够在含有cre调控的cas9等位基因(h11lsl-cas9)、荧光cre报告等位基因(r26lsl-tomato)以及熟知的肿瘤抑制基因p53(p53flox)或lkb1(lkb1flox)的floxed等位基因的小鼠中进行肿瘤启动。我们使用aav8衣壳来包装aav-krashdr/sgkras/cre文库,该衣壳可实现高滴度产生、小鼠肺上皮细胞的体内高效转导(图28)以及各种成体小鼠组织的转导35。我们用aav-krashdr/sgkras/cre来初始转导培养中的表达cas9的细胞,以确定aav/cas9介导的hdr是否为将点突变工程改造至内源性kras基因座中的无偏差方法(图27e)。krashdr特异性pcr扩增,然后对转导细胞进行高通量测序确认了所有点突变型kras等位基因的产生(图27f、g)。此外,体外krashdr等位基因频率与它们在aav-krashdr/sgkras/cre质粒文库中的呈现相关联。该结果证实,使用我们的aav载体的hdr不会通过krashdr模板中的任何单核苷酸kras密码子12或13点突变产生可辨识偏差。因此,具有特定kras突变型等位基因的肿瘤的任何差异扩增可以归因于kras变体之间的生物化学差异,而非使用具有每个kras等位基因的供体dna模板的hdr的效率差异(图27h)。为了确定体细胞中的hdr是否可以启动肿瘤,以及研究kras变体驱动肿瘤发生的能力是否有所不同,我们将aav-krashdr/sgkras/cre文库气管内递送至具有h11lsl-cas9等位基因的小鼠的肺(图24和图29)。具体而言,我们转导了三种不同基因型的小鼠,从而提供对肿瘤抑制基因的同步失活是否能够调节kras变体致癌性的见解:1)rosa26lsl-tomato;h11lsl-cas9(t;h11lsl-cas9)小鼠,2)p53flox/flox;t;h11lsl-cas9(pt;h11lsl-cas9)小鼠,其中病毒启动的肿瘤缺乏p53,以及3)lkb1flox/flox;t;h11lsl-cas9(lt;h11lsl-cas9)小鼠,其中病毒启动的肿瘤缺乏lkb1(图24a和图29a)。lt;h11lsl-cas9小鼠首先显示出肿瘤发展的迹象,包括在aav施用后大约五个月的呼吸急促和体重下降。这与具有cre调控的krasg12d等位基因和lkb1丧失的小鼠中的肺部肿瘤的快速生长一致。lt;h11lsl-cas9小鼠具有很多原发性肺部肿瘤产生的非常高的肿瘤负荷(图24b、c和图29b-d)。这些小鼠的肺部的组织学分析证实了大型腺瘤和腺癌的存在(图24b和图29b)。pt;h11lsl-cas9小鼠还产生了许多大型原发性肺部肿瘤。与lt;h11lsl-cas9小鼠相比,在pt;h11lsl-cas9小鼠中启动的肿瘤具有更明显的核异型,这是p53缺陷的特征。最后,即使在较晚的时间点,t;h11lsl-cas9小鼠也产生较小、进展较少的病灶(图24b、c和图29b-d)。以低10倍的aav-krashdr/sgkras/cre剂量转导的小鼠产生的肿瘤成比例减少(图29e)。用aav-krashdr/sgkras/cre转导的若干lt;h11lsl-cas9和pt;h11lsl-cas9小鼠还产生浸润性原发性肺部肿瘤、在胸膜腔中产生弥散性肿瘤细胞(dtc)以及淋巴结转移(图24d、e和图29f、g)。因此,aav-krashdr/sgkras/cre诱导的肿瘤可以进展为恶性和转移性肺癌。我们通过用1:10000稀释度的aav-krashdr/sgkras/cre来感染kraslsl-g12d;pt和kraslsl-g12d;lt小鼠,从而估算了肺中aav/cas9介导的体细胞hdr的效率,以使得致癌性krasg12d将在所有病毒转导的细胞中表达。这些小鼠产生的肿瘤为其中致癌性kras等位基因由aav/cas9介导的体细胞hdr产生的小鼠的大约一半。该结果与在0.02%和0.1%之间的hdr频率一致,从而使得能够在单独的小鼠中平行稳健地启动多个肺部肿瘤(图24c)。重要的是,将不含sgkras(aav-krashdr/cre)的类似载体文库递送至t、pt和lt小鼠不会导致有效的肿瘤启动,这表明p53缺陷和lkb1缺陷二者与高水平aav载体转导组合都不足以驱动肺部肿瘤发生(图24c和图30)。为了验证使用aav-krashdr/sgkras/cre启动的肿瘤具有突变型krashdr等位基因,我们分析了来自lt;h11lsl-cas9和pt;h11lsl-cas9小鼠的大型单独肺部肿瘤的facs分离的tomato阳性癌细胞中的kras基因座。使用krashdr等位基因特异性引物进行的pcr扩增证实了具有唯一条形码的致癌性kras等位基因在每个肿瘤中的存在(图24f和图31a、b)。有趣的是,尽管初始aav文库中不存在任何可检测的hdr偏差以及突变型等位基因的相对统一呈现,在约50个大型肺部肿瘤中仅鉴定到十三个kras变体中的五个(图24f)。该结果与肺部肿瘤发生中的kras变体的差异选择一致。通过分析单独的肿瘤,我们能够谨慎评估肿瘤细胞中存在的krashdr等位基因以及第二kras等位基因二者(图31)。大约一半致癌性krashdr等位基因来自完美hdr事件,其中kras点突变和唯一条形码无缝地重组到内源性kras基因座中。其余的krashdr等位基因从5’末端至突变型外显子2是无缝的,但是在内含子2中含有小重复、插入或缺失(图31d)。重要的是,预期这些改变都不能破坏从突变型外显子2至外显子3的剪接。另外,几乎所有肿瘤都在第二kras等位基因中具有cas9诱导的插入缺失,这与致癌性kras驱动的人肿瘤中野生型kras等位基因的频繁丧失一致(图31e、f)。虽然此前的研究已经记录了在小鼠中野生型kras等位基因失活后krasg12d和krasq61l驱动的肺部肿瘤生长得到增强,但是我们的结果表明很多致癌性kras变体在肺部肿瘤生长期间可能受到野生型kras的抑制。除驱动人肺癌之外,致癌性kras几乎也是人胰管腺癌(pdac)中遍在的。krasg12d或krasg12v的表达以及p53的失活会导致小鼠模型中pdac的发展。为了确定aav/cas9介导的体细胞hdr是否也可以在胰腺上皮细胞中诱导癌症启动的致癌性点突变,我们通过逆行胰管注射用aav-krashdr/sgkras/cre转导pt;h11lsl-cas9小鼠(图25a和图32a)。这些小鼠产生癌前胰腺上皮内瘤变(panin)以及pdac(图25b和图32b、c、f)。几只小鼠还产生浸润性和转移性pdac,这与人疾病的侵袭性质一致(图25c和图32d-f)。来自几个大型胰腺肿瘤块的krashdr等位基因的测序发现了具有唯一条形码的致癌性kras等位基因(图24d)。有趣的是,虽然只分析了四种样品,但是只观察到krasg12d和krasg12v—人胰腺癌症中的两个最常见的kras突变。与致癌性kras启动pdac的要求一致的是,通过逆行胰管注射我们的阴性对照aav-krashdr/cre载体来将胰腺细胞转导至pt小鼠中不会诱导任何胰腺肿瘤(图32f)。人软组织肉瘤也经常在ras途径以及tp53中具有突变。已经通过krasg12d的表达和p53失活在基因工程小鼠中诱导了肉瘤。为了确定aav/cas9介导的体细胞hdr是否可以用于将点突变引入kras和驱动肉瘤形成,我们向pt;h11lsl-cas9小鼠的腓肠肌进行aav-krashdr/sgkras/cre的肌肉内注射(图25e和图33a)。这些小鼠产生快速生长和浸润性肉瘤,所述肉瘤含有具有唯一条形码的krasg12d、krasg12a和krasg13r等位基因(图25f-h和图33)。该平台在不同组织中的肿瘤发生建模—从启动至恶性进展—中的成功应用,突出显示了其在多种癌症类型中在致癌性驱动子突变的多重功能分析方面的广泛适用性。虽然目前评估自身癌症模型中的基因功能的方法大大依赖于肿瘤数量和大小的人工定量,但是我们建立了将肿瘤细胞数量与直接来自成批组织的肿瘤基因型联系起来的简单、高通量、多重方法(图23d和4a)。由于通过hdr引入体细胞的唯一dna条形码将随着细胞的分裂而使细胞数量增加,因此可以通过条形码区的深度测序来确定给定肿瘤中的相对癌细胞数。此外,可以通过在深度测序之前将归一化对照添加至每个样品来估算每种肿瘤中细胞的绝对数。为了确定基因型和估算用aav-krashdr/sgkras/cre转导的t;h11lsl-cas9、pt;h11lsl-cas9和lt;h11lsl-cas9小鼠的整个肺的每个肿瘤中的绝对癌细胞数,我们首先将具有已知条形码的5x105个细胞的dna添加至每个样品(图26a和图34)。然后我们从成批肺部样品提取dna,pcr扩增krashdr等位基因并且对每个等位基因的变体-条形码区进行深度测序(图23d和4a以及图34)。在高通量测序后,我们校正了反复出现的测序误差以及具有相同的条形码的单独肿瘤的可能性。然后我们通过将肿瘤条形码测序读段计数归一化为来自归一化对照dna的读段数来估算每个肿瘤中的绝对癌细胞数。该分析流程具有极高的可重复性,并且在技术重复中肿瘤大小高度一致(图35)。通过对来自成批组织的单独肿瘤进行平行定量分析,这种基于hdr的条形码编制和深度测序方法提供了前所未有的体内肿瘤景观图。krashdr变体-条形码区的高通量测序在t;h11lsl-cas9、pt;h11lsl-cas9和lt;h11lsl-cas9小鼠中发现了很多aav-krashdr/sgkras/cre诱导的肺部肿瘤(图36a-c)。将肿瘤数量归一化为aav-krashdr/sgkras/cre载体文库中每个krashdr等位基因的初始呈现允许我们直接比较每个kras变体的体内致癌性(图26b和图36d、e)。在超过500种肿瘤中,krasg12d是最常见的变体,这与krasg12d是非吸烟者的人肺部腺癌中最常见的kras突变一致。krasg12a、krasg12c和krasg12v(krasg12d之后人肺部腺癌中最常见的kras变体)以及krasg13s被鉴定为肺部肿瘤发生的中驱动子,但是与krasg12d相比存在于显著较少的肿瘤中(图26b)。有趣的是,krasg12r和krasg13r也被鉴定为有效致癌性变体,尽管在人肺癌中很少发生突变(图26b)。我们在pt;h11lsl-cas9和lt;h11lsl-cas9小鼠中启动了肿瘤,以直接评估同步肿瘤抑制物改变是否能够调节不同的kras变体启动和驱动肿瘤生长的能力。有趣的是,虽然kras致癌性的总体谱随着lkb1失活显著改变,但是我们未观察到单独的kras变体在p53或lkb1同时失活的肿瘤中的相对肿瘤发生潜力的显著差异(图26c-e和图36)。该数据与其中这些致癌性kras变体体内诱导的信号传导强度不足以参与p53途径的模型一致;因此,虽然p53的作用约束了肿瘤进展,但是它不限制具有某些kras基因型的肿瘤的初始扩增。另外,虽然lkb1缺陷增加了肿瘤生长,但是lkb1缺陷诱导的信号传导不会优先与kras的特定突变形式诱导的下游信号协同作用。由于我们的肿瘤条形码编制和测序平台允许我们平行鉴定很多来自成批肺部的单独肺部肿瘤,因此我们预计,我们也可以使用该方法来克服鉴定和分析人pdac的自身小鼠模型启动的多病灶肿瘤块中的单独的胰腺肿瘤克隆的挑战31。因此,我们还分析了来自用aav-krashdr/sgkras/cre转导的pt;h11lsl-cas9小鼠的成批胰腺肿瘤样品(图26f和图37a、b)。胰腺肿瘤块的条形码测序发现了每只小鼠的多个原发性肿瘤克隆,每个克隆具有含有kras密码子12或13中的点突变和唯一dna条形码的krashdr等位基因。胰腺肿瘤展示了致癌性kras等位基因偏好,其中krasg12d、krasg12v和krasg12r是最普遍的变体(图26f)。值得注意的是,这三个kras变体也是人pdac中最普遍的致癌性kras突变。除了确定特定kras变体的体内致癌性之外,我们的条形码测序方法还允许我们鉴定来自pdac块的多区域测序的邻接的肿瘤克隆,并发现原发性肿瘤及其转移后代之间的克隆关系(图26g和图37c)。突变在人癌症中的盛行与突变发生的频率和突变驱动肿瘤发生的程度二者有关。通过使用aav/cas9介导的体细胞hdr将点突变以无偏差方式引入内源性kras基因座中,我们确定kras变体在数量上具有不同的驱动肺部肿瘤发生的能力(图4b和图36)。此外,使用我们的基于hdr的方法在小鼠中启动的胰腺肿瘤展示了针对相同的显性kras变体作为人pdac的选择,这表明在人pdac中观察到的kras突变谱可能由kras突变体之间的生物化学差异而不是它们的突变率的差异驱动(图26f和图37)。为了开始理解每个kras变体的生物化学性质如何影响其体内致癌性,我们研究了此前记录的kras变体的生物化学行为之间的关系,以及它们在我们的研究中驱动肺部或胰腺肿瘤形成的能力(图38)。值得注意的是,虽然kras突变导致了被认为对kras功能关键的生物化学特征的显著差异(例如,gtp酶活性和raf激酶亲和力),但是单独的生物化学性质不能预测体内kras变体致癌性。该结果表明,kras变体的体内致癌性可以由替代生物化学性质,或者更可能,通过多个生物化学输出的整合最好地描述。这项工作突出显示了,我们的aav/cas9介导的体细胞hdr方法是一种可用于多组突变的体内致癌性的成本效益好的并且系统性平行研究的定量、可调整、模块化方法。可实现kras功能的体内遗传解剖的多重方法代表了正在进行的ras蛋白的突变形式的生物化学和细胞培养研究的关键补充。该方法将实现对普遍的癌基因的多种突变以及很多常见癌症类型中的罕见、推定致癌性突变的功能的前所未有的理解。最后,我们设想该平台将显著加速针对精确定义的癌症遗传亚型的靶向疗法的发现和临床前验证。方法靶向kras的sgrna的设计、产生和筛选为了获得增强小鼠体细胞中的同源定向修复(hdr)的靶向kras的sgrna,我们鉴定了所有可能的靶向kras外显子2的20-bpsgrna(使用共有cas9pam:ngg)以及侧接的内含子序列,并且使用可用的sgrna设计/评分算法对它们进行预测中靶切割效率评分。然后我们凭经验确定了三个靶向kras的sgrna的切割效率(sgkras#1:gcagcgttacctctatcgta;sgkras#2:gctaattcagaatcactttg;sgkras#3:gactgagtataaacttgtgg)(图27a)。简而言之,针对上文所述的每个靶向kras的sgrna产生lenti-u6-sgrna/cre载体。定点诱变(neb)被用于将sgrna插入亲本慢病毒载体,该载体含有驱动sgrna转录的u6启动子以及驱动pgk启动子的cre-重组酶。经由用每种lenti-sgkras/cre病毒转导培养中的lsl-yfp;cas9细胞来确定每种sgkras的切割效率。在感染(转导)后48小时我们通过facs来分离yfp阳性细胞,提取dna,pcr扩增被靶向的kras基因座(正向引物:tcccctcttggtgcctgtgtg;反向引物:aagcccttcctgctaatctcggag)并对扩增子进行桑格测序(测序引物:gcacggatggcatcttggacc)。通过tide分析测序迹线,以确定插入缺失诱导的百分比。由于全部三个sgrna均在预期基因座处诱导插入缺失,因此靶向最接近kras密码子12和13的序列的sgkras(sgkras#3)被用于所有后续实验,因为预期这最有利于所需基因座处的hdr(图27a)。aav-krashdr质粒文库的设计、构建和验证aav-krashdr/sgkras/cre骨架的产生用聚合酶(neb)来pcr扩增来自pll3.3;u6-sgkras/pgk-cre的u6-sgkras/pgk-cre盒,进行topo克隆(invitrogen),并通过测序进行验证。为了产生aav-sgkras/cre载体,使用xhoi/spei来移除388-mcsaav质粒骨架的itr之间的序列。用xhoi/xbai从topo载体消化u6-sgkras/pgk-cre盒,并将1.9-kb片段连接至xhoi/spei消化的388-mcs骨架,从而破坏spei位点。在mlui消化后将bghpolya序列插入cre的3’。从基因组dnapcr扩增鼠科动物kras的外显子2周围的约2-kb区域(正向引物:gccgccatggcagttcttttgtatccatttgtctctttatctgc;反向引物:gccgctcgagctcttgtgtgtatgaagacagtgacactg)。随后将扩增子克隆至topo载体(invitrogen)。使用定点诱变(neb)将avrii/bsiwi位点引入topo克隆的2-kbkras序列(avrii正向引物:tgagtgttaaaatattgataaagtttttg;avrii反向引物:cctaggtgtgtaaaactctaagatattcc;bsiwi正向引物:cttgtaaaggacggcagcc;bsiwi反向引物:cgtacgcagactgtagagcagc;限制性位点以下划线表示,错配碱基以小写表示)。用ncoi/xhoi从topo释放具有avrii/bsiwi位点的kras片段,并连接至ncoi/xhoi消化的aav-sgkras/cre,以产生aav-krashdr/sgkras/cre骨架。aav-krashdr/cre骨架的产生为了产生不具有靶向kras的sgrna的aav-krashdr骨架,用noti/xbai从topo克隆切下pgk-cre,并连接至noti/xbai消化的388-mcsaav质粒骨架。如上文所述添加bghpolya序列和小鼠kras片段,以产生对照aav-krashdr/cre骨架。多种kras变体/条形码区域的设计和合成为了将激活的单独点突变的文库和dna条形码引入aav骨架的krashdr序列,我们合成了四个295-bpkras片段,这些片段在kras密码子12和13的前两个碱基对中的每个处具有简并“n”碱基(a、t、c或g)(integrateddnatechnologies)(图27b)。根据设计,四个片段库中的每个由密码子12和13处的三个非同义、单核苷酸突变,以及作为对照的野生型kras序列组成。因此,由于四个库中的每个含有野生型片段,预期野生型kras等位基因的总体呈现比突变型kras等位基因中的每个大大约四倍。合成片段还含有sgkras靶序列中的沉默突变和相关的原间隔序列邻近基序(pam*),对于单独肿瘤条形码编制,通过将简并碱基引入下游kras密码子的摇摆位置来产生八个核苷酸的随机条形码(图27b)。最后,每个片段包含侧接的avrii和bsiwi限制性位点,用于克隆至aav-krashdr骨架(图27b)。kras突变体/条形码片段向aav-krashdr载体的连接以等比率组合四个合成片段库,并进行pcr扩增(正向引物:cacacctaggtgagtgttaaaatattg;反向引物:gtagctcactagtggtcgcc)。用avrii/bsiwi消化扩增子,通过乙醇沉淀来纯化,并连接至两个aav-krashdr骨架(图27c)。将每个连接的质粒文库转化至stbl3电感受态细胞(neb),并接种至20个lb-amp平板,所述平板产生约3x105个细菌菌落/文库。将菌落刮入lb-amp液体培养基中,并在37℃下扩增六小时,以增加质粒收率,从而获得足够的用于aav产生的质粒dna。然后使用maxiprep试剂盒(qiagen)从细菌培养物提取质粒dna。aav-krashdr质粒文库的验证为了确定每个aav质粒文库中每个kras变体的呈现和条形码核苷酸的分布,用以含有多重标签(以下划线表示的n)的illumina衔接子(小写)结尾的引物来pcr扩增纯化的aav质粒文库(正向引物:aatgatacggcgaccaccgagatctacactctttccctacacgacgctcttccgatctctgctgaaaatgactgagtataaactagtagtc;反向引物:caagcagaagacggcatacgagatnnnnnngtgactggagttcagacgtgtgctcttccgatcctgccgtcctttacaagcgtacg),然后在miseq上进行深度测序。aav衣壳血清型肺上皮细胞转导分析使用ca3(po4)2三重转染方案,用pad5辅助蛋白、ssaav-rsv-gfp转移载体、以及针对所关注的九个衣壳中的每个的假型化质粒:aav1、2、3b、4、5、6、8、9_hu14和dj来产生重组aav-gfp载体。在hek293t细胞(atcc)中产生病毒,然后进行双氯化铯密度梯度纯化和透析,如上文所述。通过taqmanqpcr来针对gfp滴定raav载体制备物(正向引物:gacgtaaacggccacaagtt;反向引物:gaacttcagggtcagcttgc;探针:6-fam/cgagggcgatgccacctacg/bhq-1)。为了鉴定针对成体肺上皮细胞转导的最佳aav血清型,每只小鼠经由气管内施用接受60μl最大滴度的假型化aav-gfp。在aav施用后5天分析小鼠。将肺分离至单细胞悬浮液,并制备用于gfp阳性细胞的facs分析,如上文所述。通过分析>10000个活设门细胞来确定gfp阳性百分比(参见图28)。aav-krashdr质粒文库的产生和滴定使用ca3(po4)2三重转染方案,用pad5辅助蛋白、paav2/8包装质粒以及上文所述的具有条形码的kras文库转移载体库来产生aav文库。在hek293t细胞中进行转染,然后进行双氯化铯密度梯度纯化和透析,如上文所述。通过taqmanqpcr来针对cre滴定aav文库(正向引物:tttgttgccctttattgcag;反向引物:cccttgcggtattctttgtt;探针:6-fam/tgcagttgttggctccaacac/bhq-1)。体外aav/cas9介导的hdr密码子12和13处的突变周围的核苷酸变化(使sgrna识别位点和pam基序突变的密码子12/13的5’的三个核苷酸变化,以及条形码序列中的最多10个变化)使以下情况不太可能发生:kras密码子12和13处的点突变将差异化影响hdr的比率。我们还是测试了hdr效率是否可能受到单独的krashdr等位基因的序列差异的影响。为了诱导体外aav/cas9介导的hdr,我们用纯化的aav-krashdr/sgkras/cre文库来转导lsl-yfp/cas9细胞(图27e)。用10μmscr7(xcessbio)(非同源末端连接(nhej)的抑制剂)将细胞维持在细胞培养物中,以促进同源定向修复。在转导后96小时,通过酚/氯仿提取然后是乙醇沉淀从lsl-yfp/cas9细胞分离dna。使用我们开发的pcr策略从该dna扩增kras基因座,用于进行整合进内源性kras基因座的krashdr等位基因的特异性扩增。然后我们对这些扩增子进行深度测序,以确定体外hdr后krashdr等位基因的呈现(参见以下“文库制备和来自成批组织的肿瘤条形码的测序”部分,以了解关于pcr和测序的细节)。小鼠和肿瘤启动lkb1flox(l)、p53flox(p)、r26lsl-tomato(t)、h11lsl-cas9和kraslsl-g12d(k)小鼠已在上文有所描述。如所描述进行通过气管内吸入的aav施用(启动肺部肿瘤)、逆行胰管注射(启动胰腺肿瘤)以及肌肉内腓肠肌注射(启动肉瘤)。在pt;h11lsl-cas9、lt;h11lsl-cas9和t;h11lsl-cas9小鼠中用60μlaav-krashdr/sgkras/cre(1.4x1012vg/ml)、在pt、lt和t小鼠中用60μaav-krashdr/cre(2.4x1012vg/ml)或在kpt和klt小鼠中用60μlaav-krashdr/sgkras/cre(1.4x1012vg/ml)(在1×pbs中1:10000稀释)来启动肺部肿瘤。在pt;h11lsl-cas9小鼠中用100-150μlaav-krashdr/sgkras/cre(1.4x1012vg/ml)或在pt小鼠中用100-150μlaav-krashdr/cre(2.4x1012vg/ml)来启动胰腺肿瘤。如果需要,将在1×pbs中1:10稀释的aav-krashdr/sgkras/cre另外施用于小鼠的肺或胰腺。在pt;h11lsl-cas9中用30μlaav-krashdr/sgkras/cre(5.2x1012vg/ml)来启动肉瘤。当小鼠表现出肿瘤发展的症状时对它们实施安乐死。斯坦福大学实验动物护理和使用委员会(institutionalanimalcare&usecommitteeofstanforduniversity)批准了所有小鼠程序。单独肿瘤的分析单独肺部肿瘤的分析携带肺部肿瘤的小鼠表现出肿瘤发展的症状,并且在病毒施用后对所述小鼠进行分析4-10个月。根据需要,通过肺部重量以及通过荧光解剖镜下宏观tomato阳性肿瘤的定量来评估肺部肿瘤负荷(单只lt;h11lsl-cas9小鼠具有受限于一个肺叶的小区域的最小tomato阳性信号,这表示aav的不当气管内施用,已从研究中移除)。在荧光解剖显微镜下,从成批肺部解剖具有不可见的多病灶的最大的单独肺部肿瘤,以用于测序。对于一些肺部肿瘤,使用斯坦福共享facs设施(stanfordsharedfacsfacility)中的facs机器(ariasorter;bdbiosciences)来纯化tomato阳性肿瘤细胞。还收集来自单独小鼠的若干肺叶,以用于组织学分析。单独胰腺肿瘤块的分析携带胰腺肿瘤的小鼠表现出肿瘤发展的症状,并且在病毒施用后对所述小鼠进行分析3-4个月。由于胰腺肿瘤在很大程度上似乎是多病灶的,解剖下含有tomato阳性肿瘤块的胰腺的单独区域,并对其进行facs纯化,以用于测序(用1:10稀释的aav-krashdr/sgkras/cre文库处理的小鼠也产生胰腺肿瘤块,所以被包括在这些分析中)。保留若干胰腺的区域,以用于组织学分析。单独肉瘤的分析携带肉瘤的小鼠具有明显的肿瘤发展,在病毒施用后对这些小鼠进行分析3-7个月。保留每个肉瘤的区域,以用于测序,并且保留邻近的区域,以用于组织学分析。单独肿瘤中kras等位基因的表征从facs纯化的肿瘤细胞提取用于测序的dna,并用dneasy血液和组织提取试剂盒(qiagen)对肿瘤样品进行去分选。为了鉴定肿瘤中的kras点突变和条形码,我们使用两种方案对krashdr等位基因进行pcr扩增和测序,已针对若干变量对这些方案进行了优化:包括退火温度、延伸时间和引物序列(方案1–正向引物:ctgctgaaaatgactgagtataaactagtagtc;反向引物:agcagttggcctttaattggtt;测序引物:aatgatacggcgaccaccgagatctacac;退火温度66℃;方案2–正向引物:gctgaaaatgactgagtataaactagtagtc;反向引物:ttagcagttggcctttaattgg;测序引物:gcacggatggcatcttggacc;退火温度64℃)。这些方案被用于特异性扩增来自单独肿瘤的整合krashdr等位基因,因为所述等位基因掺入了与密码子12和13上游的pam区域中的工程改造突变重叠的正向引物,以及同源臂外部的反向引物。长延伸时间(2-3分钟)被用于扩增所有krashdr等位基因,甚至是在kras基因座的内含子2中含有插入或重复那些等位基因(图31d)。在通过nhej而不是hdr进行dna修复后,除了经由hdr将所需的点突变引入内源性kras基因座之外,预期使用crispr/cas9来靶向kras外显子2还会导致切割位点处的插入缺失。为了表征这些修饰,我们使用通用pcr方案来扩增两个kras等位基因(正向引物:tcccctcttggtgcctgtgtg;反向引物:ggctggctgccgtcctttac;测序引物:caagctcatgcgggtgtgtc;退火温度72℃)。通过该方法鉴定dna切割位点处的插入和缺失谱(图31e、f)。对于一些单独的肿瘤样品,在上述pcr和测序策略之后,两个kras等位基因的序列并不特别明显。topo克隆(invitrogen)和转化来自这些样品的pcr产物,质粒制备每个样品的若干菌落并测序,以表征每个肿瘤中的两个kras等位基因。在生物学重复和技术重复二者中,该方法是可重现和可靠的,并且鉴定了许多hdr诱导的致癌性kras等位基因。在约50个肿瘤中鉴定出含有插入缺失的kras等位基因(图31a、b)。这些分析还发现了来自单独的肺部肿瘤的一些kras等位基因的若干其他意外特征。在所分析的单独肺部肿瘤的小亚组中观察到密码子24处的三个独特错义突变(i24l、i24n、i24m)。这些改变(如果有的话)的功能未知。此外,我们最初预计,krashdr模板向内源性kras基因座的重组将出现在被工程改造至krashdr模板中的avrii和bsiwi位点的外部(图31c)。然而,通过改变外显子2上游的2个碱基对97个碱基对来工程改造的avrii位点在25个肿瘤中的5个中不存在,其中我们直接分析了krashdr等位基因的该区域。通过改变外显子2下游的1个碱基对20个碱基对来工程改造的bsiwi位点在58个肿瘤中的11个中不存在。这些发现表明,虽然krashdr模板的重组最常出现在更大、更远的同源臂内,但是它还以可检测的频率出现在5’和3’错配侧接的非常短的同源性区域中(包括pam*突变、kras密码子12或13突变以及条形码中的8个潜在错配)。在我们最初鉴定到krashdr等位基因中的重复存在于一些肿瘤之后,我们设计了pcr引物来特异性扩增出现在hdr整合的kras基因座的任一侧的krasl外显子2的重复(右手重复–正向引物:tgaccctacgatagaggtaacg;反向引物:ctcatccacaaagtgattctga;测序引物:tgaccctacgatagaggtaacg;左手重复–正向引物:tgagtgttaaaatattgataaagtttttg;反向引物:tccgaattcagtgactacagatg;测序引物:tgagtgttaaaatattgataaagtttttg)。这些重复特异性pcr方案中的每个以相反的方向使用邻近引物对,确保扩增仅出现在重复存在的情况下。鉴定到不同长度的重复(图31d),包括野生型外显子2的后半部分或整个外显子2的重复(但是缺乏剪接接受体的关键区域)。还观察到内含子2的区域的缺失和重复。此外,我们还观察到aav载体的部分(包括u6启动子和病毒itr)整合到内含子2中。鉴于这些改变的大小和位置,预计不会改变kras突变型外显子2至外显子3的剪接,这与致癌性kras驱动肿瘤发生的要求一致。具有已知krashdr等位基因和条形码的细胞系的高通量测序的归一化对照的产生为了建立用作测序归一化对照的细胞系,将单个大型肿瘤从pt;h11lsl-cas9小鼠解剖下,消化至单细胞悬浮液中,并接种以产生细胞系。在扩增这些细胞然后提取dna之后,pcr扩增kras外显子2(正向引物:tcccctcttggtgcctgtgtg;反向引物:ggctggctgccgtcctttac)。对pcr产物进行测序(使用上文所述的特异性和通用测序引物),以确认krashdr等位基因和条形码的存在。鉴定出具有唯一条形码(cgggaagtcggcgcttacgatc)的单个krasg12v等位基因。来自该细胞系的基因组dna被用作所有成批肺部样品的高通量测序的归一化对照(图34)。成批组织处理和dna提取成批肺部组织处理和dna提取从感染(转导)的小鼠解剖下成批肺部样品,并且在处理前储存在-80℃下。为了提取用于测序的dna,将样品解冻并转移至50ml锥形管中。将20ml裂解缓冲液(100mmnacl、20mmtrisph7.6、10mmedtaph8.0、0.5%sds,溶于h2o)和200μl蛋白酶k(20mg/ml)添加至每个样品。然后,将3μg(约5x105个基因组)归一化对照dna添加至每个样品(图25a和图35b)。然后使用组织匀浆机小心地对样品进行匀浆,在每个样品之间使干净的10%漂白剂、70%乙醇和1×pbs通过来清洗组织匀浆机。将匀浆的样品在55℃下裂解过夜。通过酚/氯仿提取然后是乙醇沉淀来从组织裂解物分离dna(图37a、b)。成批胰腺组织处理和dna提取解剖、消化胰腺肿瘤块,并通过facs分离活细胞(dapi阴性)、谱系(cd45、cd31、ter119、f4/80)阴性细胞、tomato阳性细胞。归一化对照不添加至胰腺癌样品。使用dneasy血液和组织提取试剂盒(qiagen)从facs分离的赘生性细胞分离dna,然后通过乙醇沉淀进一步纯化。从成批组织进行文库制备和肿瘤条形码测序为了以大规模平行和定量方式发现具有每个kras变体的肿瘤的数量和大小,我们开发了可实现具有条形码的krashdr等位基因的多重测序的两轮pcr策略(图27f)。对于第1轮pcr,我们使用与含有三个pam和sgrna靶位点突变的krashdr序列互补的正向引物(pam*;第1轮正向引物序列以粗体表示)(第1轮正向引物:gctgaaaatgactgagtataaactagtagtc)(seqidno:2)和与aav-krashdr/sgkras/cre载体中的hdr模板中不存在的内源性kras基因座的下游区域互补的反向引物(第1轮反向引物:ttagcagttggcctttaattgg)(seqidno:3)。选择该引物对以特异性扩增基因组krashdr等位基因,但不扩增从成批携带肿瘤的组织纯化的dna中存在的高丰度的野生型kras等位基因或潜在的附加体aav-krashdr/sgkras/cre载体。另外,p5衔接子(以斜体表示)、8-bp自定义i5索引(n)和测序引物序列(读段1)(以下划线表示)被包括在第1轮正向引物的5’末端,以进行多重测序(illumina测序的第1轮正向引物:aatgatacggcgaccaccgagatctacacnnnnnnnnacactctttccctacacgacgctcttccgatctgctgaaaatgactgagtataaactagtagtc)(seqidno:4)。重要的是,由于单独的肿瘤中krashdr等位基因的表征发现了在kras内含子2中产生多种插入缺失的hdr的一些差异(图32d),在第1轮pcr中只进行4个(肺部样品)或6个(胰腺样品)循环,以使在不同长度的产物的扩增期间产生偏差的可能性最小化。此外,将高效聚合酶(热启动高保真聚合酶,neb;64℃退火温度)和长延伸时间(3:00分钟)用于确保所有krashdr等位基因的稳健扩增。使用4个和40个之间的单独100μlpcr反应来扩增肺部基因组dna中的krashdr等位基因,然后在扩增后合并以减少pcr累积的作用(图34a)。这些100-μlpcr反应中的每个都含有4μgdna模板,以从krashdr等位基因的大型初始库进行扩增。在第1轮扩增后,合并所有重复pcr反应并使用qiaquickpcr纯化试剂盒(qiagen)来清洗100μl每个样品。将纯化的第1轮pcr扩增子用作100μl第2轮文库pcr的模板dna(热启动高保真聚合酶,neb;72℃退火温度;对于肺部样品35个循环,对于胰腺样品40个循环)。第2轮pcr扩增第1轮pcr扩增子中存在的kras外显子2序列中的整个112-bp区域。第2轮反向引物在5’末端含有p7衔接子(以斜体表示)、反向互补的8-bp自定义i7索引(“n”)和反向互补的illumina测序引物序列(读段2)(以下划线表示),以对illumina文库进行双索引、双末端测序(第2轮反向引物#1:caagcagaagacggcatacgagatnnnnnnnngtgactggacttcagacgtgtgctcttccgatccgtagggtcatactcatccaca)(seqidno:5)。第2轮pcr正向引物与p5illumina衔接子互补,该衔接子在第1轮pcr期间通过正向引物添加至扩增的krashdr等位基因(第2轮正向引物:aatgatacggcgaccaccgagatctacac)(seqidno:6)。该引物被用于扩增第1轮pcr扩增子,但不扩增可以从第1轮pcr反应携带下来的任何污染的基因组dna。此外,将编码p7衔接子序列的第二反向引物以与另外两个引物相同的浓度添加至第2轮pcr反应(第2轮反向引物#2:caagcagaagacggcatacgagat)(seqidno:7)。该引物结合反向互补的p7衔接子序列,该序列通过第2轮反向引物#1添加至krashdr扩增子。由于第2轮pcr进行35-40个循环,添加p7衔接子(第2轮反向引物#2)以限制较长的第2轮反向引物#1所产生的非特异性扩增的量。在第2轮扩增后,在2.5%琼脂糖凝胶上运行100-μlpcr反应,并切下预期大小的条带。使用qiaquick凝胶提取试剂盒(qiagen)从凝胶片段提取dna。使用生物分析仪(agilent)来测定纯化的文库的质量和浓度。然后将具有唯一的双索引的单独的文库合并在一起,以使得与来自具有更低的肿瘤负荷的小鼠的那些文库相比,最初来源于具有更高肿瘤负荷的小鼠的文库在最终库中以更高的比率呈现(图34a)。将总共35个单独的样品组合成两个文库。在生物分析仪(agilent)上确认每个库的质量和浓度。然后在hiseq泳道上使用多重150bp双末端快速运行测序程序(elimbiopharmaceuticals)对每个最终文库进行深度测序。估算具有条形码的肿瘤的大小和数量的illumina测序数据的分析我们开发了从我们的解多重测序数据识别肿瘤的流程。该流程使用被设计为使扩增子的深度测序数据降噪的算法(dada2),采用唯一条形码序列,并除去反复出现的测序误差。我们对该算法进行了定制,以使假性肿瘤识别的出现最小化,并且使技术偏差(包括读段深度差异、测序机器误差率差异和条形码多样性差异)最大化。下文对该流程进行了描述,包括在aav/cas9介导的体细胞hdr驱动的肿瘤发生后对肿瘤基因型和条形码的分析的修改。合并、过滤和修剪双末端读段虽然我们的测序文库含有较小的krashdr等位基因的112-bp片段,但是我们对这些片段进行了150bp双末端测序,并合并了重叠的正向和反向读段,以降低kras密码子12和13以及krashdr等位基因的条形码区中的测序误差的可能性。使用pandaseq对重叠的双末端读段进行合并、质量过滤和修剪(片段长度:60bp;正向修剪引物:atgactgagtataaact;反向修剪引物:ctcatccacaaagtga)。识别唯一的肿瘤即使在合并正向和反向读段以减少测序误差之后,仍可检测到平均约1个误差/10000个碱基,这大概是来自反复出现的测序误差(或很少来自反复出现的pcr误差)。鉴于存在这种误差率,我们预期来自含有单核苷酸错配的大型具有唯一条形码的肿瘤的读段将被识别为大小为大型真实肿瘤的约1/10000的小型假性肿瘤。用肉眼即可看出这种现象,因为我们观察到小约3-4个数量级并且相对于特定小鼠中的最大的肿瘤含有1个核苷酸偏差的假性“肿瘤”的小聚类。另外,每个krashdr变体-条形码对还在致癌性密码子12或13中的突变型碱基中产生反复出现的测序误差。为了准确识别肿瘤,我们开发了用于通过以下步骤分析肿瘤条形码测序数据的计算和统计学流程:使用dada2来训练来自读段的非条形码区的误差模型并且将唯一的读段堆积聚类到肿瘤中我们估算了来自kras密码子12上游的7个核苷酸和最终条形码碱基下游的7个核苷酸的测序/pcr误差的残留率。然后我们使用我们的测序误差模型经由dada2将唯一的读段堆积(截短至7个核苷酸以内的具有条形码的碱基)聚类到唯一的肿瘤。使用聚类的唯一起源的最小置信度0.01(即,omega_a=0.01)。较大的阈值增加了在小鼠样品中识别的唯一肿瘤的数量。我们选择了这个较大的值,因为双末端测序似乎使我们更有把握,即唯一的读段堆积是真实的独特肿瘤。例如,我们发现该阈值消除了所有意外的读段序列(例如,在条形码外部具有不适当的核苷酸的读段),并且该阈值识别了在生物学重复之间更一致的每只小鼠中的病灶总数。这些是重要的考虑因素,因为如果不适当处理读段误差,则所识别的肿瘤数可以与测序读段深度正相关。最后,我们移除了具有与大10000×的病灶偏离仅1个核苷酸的dna序列的任何肿瘤。这仅影响1.56%的肿瘤识别。将读段堆积归一化为归一化对照以获得近似的肿瘤大小在产生读段堆积和进行上文所述的校正之后,我们将来自每个识别的肿瘤的读段数归一化为来自归一化对照的读段数,所述归一化对照在从成批携带肿瘤的组织裂解物进行dna提取之前加标至每个样品。这允许我们产生每个肿瘤中的细胞数的合理估算值,并且允许我们合并来自相同基因型和处理的小鼠的数据。然而,有若干因素会影响我们准确定量每个肿瘤中的细胞绝对数的能力。首先要考虑的是,单独肿瘤中的一些krashdr等位基因在用于测序的pcr引物内部的kras内含子2中具有插入或缺失。虽然不同大小的扩增子的存在可以产生pcr偏差,但是我们尝试通过在第1轮illumina文库pcr中仅进行4-6个循环,使用长延伸时间(约3分钟)以及使用快速(20-30秒/kb)高保真聚合酶(neb)来减小这些偏差。由于在所有样品中第2轮扩增中的最终文库pcr产物较短并且是均一的,因此在该步骤中pcr扩增不应产生偏差。此外,鉴于kras变体和条形码被敲入内源性kras基因座中,在一些肿瘤中对该区域进行基因组扩增是可能的(这已经在肺癌的小鼠模型中启动的krasg12d驱动的肺部肿瘤中有所记录)。虽然kras扩增通常不会产生非常高的kras拷贝数,但是任何扩增都会导致具有扩增的krashdr等位基因的肿瘤中的细胞数的略微高估,因为我们的从读段计数至细胞数的转换假设以下事实:每个细胞含有单个拷贝的具有条形码的krashdr等位基因。最后,归一化对照本身从kras内含子2中具有已知重复的肿瘤的细胞产生,所述肿瘤与不具有重复的肿瘤相比,在第1轮illumina文库制备中产生较大的pcr产物。因此,在归一化对照中偏离kras等位基因的任何pcr偏差都会产生不具有重复的肿瘤的大小的系统低估。估算条形码重叠率并校正肿瘤大小分布在hi-seq快速运行的两个泳道上进行的35个样品的肿瘤条形码的测序,与我们的分析流程组合,能够检测具有覆盖超过五个数量级的读段计数的唯一条形码。因此,这种方法的前所未有的分辨率允许检测成批组织中的大病灶以及小增生。然而,检测成批组织中的大量病灶的能力增加了条形码冲突的概率:两个或更多个具有相同的dna条形码的病灶出现在相同的小鼠中。条形码冲突会夸大所观察的肿瘤的大小,因为两个小“冲突”肿瘤将被鉴定为单个较大的肿瘤。因此,我们开发了条形码冲突的统计模型,以确保该问题不严重,并且不会使所识别的肿瘤的估算大小产生明显偏差。我们的条形码冲突模型说明了在我们的研究中观察针对每个kras变体的24,576个可能的条形码中的每个i的可能性pi。我们的库中的条形码频率之间的大部分可重现的差异来源于每个摇摆碱基处的核苷酸频率的统计学上独立的差异(即库中的每个条形码都不可能相同,因为在条形码片段的合成期间核苷酸浓度具有微小差异)(图23g)。因此,我们估算了条形码中每个碱基b处的每个核苷酸n的独立频率fb,n,并且使用该表根据每个条形码的序列bi,b,n来预测条形码可能性(其中如果条形码i在位置b处具有核苷酸n,则b为1,否则为0):这里,矩阵符号用于表示点积。该模型仅用21个自由参数即可预测每个条形码的频率。因为肺部样品中仍然存在一些条形码的残留过度呈现,所以在校正核苷酸频率之后,我们从所有的肺部分析简单地丢弃了10%最常观察到的条形码。在病毒产生之前,通过我们的aav-krashdr/sgkras/cre质粒库的测序(miseq)鉴定出与我们的小鼠实验无关的这些最常观察到的条形码。在该处理后,我们将∑ipi重新归一化为一。然后,我们假设每只小鼠中每个条形码的出现是多项式采样过程。针对每只小鼠中的每个观察的条形码的冲突平均数ci为:这里,μi表示每只小鼠中的条形码的平均数,而n表示肿瘤的总数(二者均未知)。使用方程n(obs)=n-σici(pi,n)和布伦特(brent)法从每只小鼠中的观察肿瘤数n(obs)来确定n。该模型发现,条形码冲突在我们的小鼠样品中通常是罕见的(平均值4.04%)。然而,冲突的可能性可以因小鼠和kras变体而变化。例如,针对wtkrashdr等位基因的冲突的平均预测数为5.8%,在一只小鼠中高达12%。预期wtkrashdr等位基因经历了最高冲突数,因为wtkras载体被刻意表示为比初始aav-krashdr/sgkras/cre质粒库中的每个突变型kras载体大约4倍(图23f)。因此,我们用每个病灶的大小除以1+ci,以使条形码冲突传递给肿瘤大小分布的偏差最小化。因为冲突是罕见的事件,所以特定小鼠中的特定冲突数可以与ci显著不同。由于该局限性,我们认为这种校正可以使条形码冲突产生的肿瘤大小分布的系统偏差最小化;然而,该校正不能有效鉴定所出现的特定冲突。确定测序质量和再现性为了确定kras变体是否具有在数量上不同的驱动肿瘤发生的能力,我们选择关注预计含有大于100000个细胞的肿瘤(即添加至来源于约5x105个细胞的每个样品的归一化对照dna“大小”的1/5)。来自重复样品(独立样品制备、测序和处理)的大于该细胞数截断值的肿瘤的回归分析展示出高度相关性(所有r2值均大于0.99;参见图36)。此外,小于该截断值的肿瘤中的估算细胞数更可能因条形码冲突和pcr扩增和测序差异而产生偏差,所有这些都会降低我们准确识别具有每个kras变体的肿瘤的大小和数量的能力。来自成批携带肿瘤的肺的测序数据分析我们通过在具有h11lsl-cas9等位基因(pt;h11lsl-cas9、lt;h11lsl-cas9和t;h11lsl-cas9)的所有小鼠基因型中对大于100000个细胞的肿瘤进行计数,并用每个变体除以在aav-krashdr/sgkras/cre质粒库中的初始呈现,从而定量了具有每个kras变体的肿瘤的相对数量(对于该分析,在移除大于第98个百分位条形码丰度的条形码之后,从与每个kras变体相关的读段总数计算每个变体在质粒库中的初始呈现;这种限制性不会明显改变结果,只是被应用于确保丰度极大的变体-条形码对不会明显影响特定变体的总体呈现)。然后对相对肿瘤数量进行换算,以使得wtkras变体具有的呈现为1。似乎来源于大于100000个细胞的肿瘤的wtkrashdr等位基因的数量相对较小。这些可以表示这样的肿瘤:其中hdr事件产生非致癌性kraswt基因型,但是由于其他原因而进化为肿瘤,或wtkras变体通过在相同的肺细胞中同时发生的hdr与致癌性kras变体一起“搭便车”,然后通过致癌性变体来驱动扩增。从成批组织解剖(并如上所述进行分析)的单独的肿瘤的少量残留细胞在我们的成批肿瘤测序数据中通常是可检测的。在肿瘤大小的所有分析中,这些解剖的肿瘤被排除在外,因为我们不能推断它们的真实大小。然而,当分析每种处理的小鼠基因型中大于100000个细胞的肿瘤的数量时,我们包括了来自单独解剖的肿瘤的数据,因为可解剖的肿瘤始终是在任何小鼠中观察到的最大肿瘤,所以肯定大于100000个细胞的阈值。使用费希尔(fischer)精确检验来确定肿瘤数量的统计学显著性差异。对于每个变体,进行两次检验,与g12d或wtkrashdr等位基因中的任一者的频率进行比较。所有p值都针对所研究的变体的数量进行了邦费罗尼校正,并且是双侧的。将双侧“多细胞”皮尔逊卡方检验用于比较相对于t;h11lsl-cas9小鼠的pt;h11lsl-cas9和lt;h11lsl-cas9小鼠中所有kras变体的肿瘤数的分布。实施例3.在体内肺部腺癌中肿瘤抑制的适合度景观癌症中存在的大多数基因组改变(单独或组合)的功能影响在很大程度上仍然不清楚。通过本文所述的实验,肿瘤条形码编制、crispr/cas9介导的基因组编辑和超深度条形码测序的整合展示出可以用于查询人肺部腺癌的自身小鼠模型中的肿瘤抑制物改变的成对组合。对31种常见肺部腺癌基因型的肿瘤抑制作用进行作图,显示出背景依赖性和差异作用强度的崎岖景观。结果癌症生长在很大程度上是多种协同基因组改变的结果。癌症基因组测序已对人癌症中的多种改变进行了编目,然而这些改变对肿瘤生长的组合作用在很大程度上尚不清楚。大多数推定的驱动子在小于百分之十的肿瘤中发生了改变,这表明这些改变可以是无活性的、无益的或仅在某些基因组背景下有益的。仅通过共存在率来推断遗传相互作用几乎是不可能的,因为可能的组合的数量可以随着候选基因数量按比例缩放。基因工程小鼠模型可以提供对在自身环境中生长的肿瘤的基因功能的见解,然而实际考虑已经阻止了对组合肿瘤抑制基因失活的广泛研究(图41)。因此,我们对体内驱动肿瘤生长的遗传相互作用的理解仍然有限。为了解决这些实际挑战,开发(本文描述)了一种使用与深度测序偶联的肿瘤条形码编制(tuba-seq)来平行定量测量很多不同的肿瘤抑制基因改变的作用的方法。tuba-seq将具有肿瘤抑制物失活(例如,crispr/cas9介导)的肺部腺癌的基因工程小鼠模型、肿瘤条形码编制和深度测序组合在一起。因为tuba-seq测量每个肿瘤的大小,并且与单独的小鼠中的多重肿瘤基因型相容,所以可以以前所未有的精度、灵敏度和通量测量生长作用。在这里,采用这种方法,对具有31种常见肿瘤抑制物基因型的致癌性krasg12d驱动的肺部肿瘤的生长进行了定量(图39)。鉴定到出乎意料的遗传相互作用,大多数肿瘤抑制物的作用被发现是背景依赖性的,并且解释了人肺部腺癌中若干遗传改变模式。在超过一半的人肺部腺癌中,肿瘤抑制物tp53是失活的。为了确定p53缺失对十种其他推定的肿瘤抑制物的生长抑制效果的作用,使用具有条形码的lenti-sgrna/cre载体(靶向很多常见肿瘤抑制基因)和四个具有条形码的lenti-sginert/cre载体(lenti-sgts-pool/cre;图39、41和42)的库在kraslsl-g12d;rosa26lsl-tdtomato;h11lsl-cas9(kt;cas9)和kt;p53flox/flox;cas9(kpt;cas9)小鼠中启动肿瘤。条形码含有两种组分,这两种组分唯一鉴定每种肿瘤及其sgrna(sgid-bc;图42)。当肺部含有广泛的增生、腺瘤和一些早期腺癌时,在肿瘤启动后15周确定每种基因型的每个肿瘤中赘生性细胞的数量。从成批携带肿瘤的肺部基因组dna扩增sgid-bc区,对产物进行深度测序,并且应用tuba-seq分析流程(本文所述)。kt;cas9和kpt;cas9小鼠的tuba-seq分析发现了针对我们的调查中的很多基因的改变的肿瘤抑制作用谱(图39和43)。通过两种此前检查的度量来汇总肿瘤大小:对数正态(ln)平均值和第95个百分位的肿瘤大小(图39)。在p53缺陷型肿瘤中,rb1、setd2、lkb1/stk11、cdkn2a或apc的失活仍然提供了生长优点,而smad4、arid1a和atm在不存在p53的情况下仅作为肿瘤抑制物出现(图39和43)。在这种背景下,另外的肿瘤抑制物的出现表明,p53缺陷增强了后续肿瘤进化。通过允许更多的突变具有适应性,p53丧失可以降低肿瘤进化的可预测性,并且有利于未来的肿瘤进化,包括治疗耐受性和转移性疾病的出现。p53的同时缺失不仅允许更多的改变具有适应性,而且还显著改变肿瘤抑制物丧失的作用程度。在kt;cas9小鼠中,rb1缺陷使肿瘤大小增加的程度小于lkb1缺陷或setd2缺陷(图39和图44a;除非另外指明,否则p<0.0001自举检验)。相比之下,在p53缺陷背景下,rb1缺陷赋予相当于lkb1缺陷或setd2缺陷的生长优点(p>0.05),这与p53和rb1肿瘤抑制物之间的强互补相互作用一致(图39)。在成批kpt;cas9肺部dna中每个靶基因座处的cas9产生的插入缺失的定量确认,具有插入缺失的lkb1、setd2和rb1等位基因的百分比相当高(图39和45)。最后,使用常规基于cre/loxp的小鼠模型确认了p53和rb1的同时失活对肺癌生长的作用(图44)。在p53完整与p53缺陷肿瘤中rb1失活的数量差异生长有益效果为研究驱动子的适合强度的改变是否可改变人肺部腺癌中的改变的频率提供了机会。实际上,rb1改变(snv和cnv)和tp53改变在人肺部腺癌中的共存在是富集的(p=0.03;图39和图44)。值得注意的是,尽管这两个改变的共存在富集约5倍,但是在校正多重假设检验之后,在所有潜在成对驱动子相互作用的初始调查中,这种相互作用在统计学上不显著,从而展示了在功能上研究共存在模式之外的遗传相互作用的需要(对于10个成对相互作用,在邦费罗尼校正之后p=0.32)。然后,通过用lenti-sgts-pool/cre在kt;lkb1flox/flox;cas9(klt;cas9)小鼠中启动肿瘤来研究lkb1和其他推定的肿瘤抑制物的组合丧失的作用(图40和43)。研究lkb1,因为它在自身模型中使肺部肿瘤生长显著增加,并且在人肺部腺癌中会经常失活(图41)。有趣的是,适应性肿瘤抑制物丧失的数量和中位数生长有益效果二者在已经快速生长的lkb1缺陷肿瘤中均被减弱(无论小鼠背景之间的统计学功效的变化如何,p<0.05,方法)。这再次表明,单个改变可以使肿瘤的整体适合度景观发生变化。适合度有益效果的普遍减弱(称为减少回报上位性)在进化中是常见的,并且表明肿瘤可以最终达到适合度平台期。apc和rb1失活是为lkb1缺陷型肿瘤提供显著生长优点的唯一改变(图40)。rb1缺陷(甚至同时发生的lkb1缺陷)增加肿瘤大小的能力,强调了rb1在细胞周期调控中的整合作用以及来自lkb1丧失的基本上不同的作用机制。apc丧失也是肺癌生长的关键驱动子,并且apc在所研究的全部三个背景下具有肿瘤抑制性。令人惊讶的是,setd2缺陷对lkb1缺陷型肿瘤的生长的作用是适度的,并且在统计学上不显著(图40)。这种冗余是惊人的,因为lkb1和setd2失活二者强烈地促进了kt;cas9和kpt;cas9小鼠的生长,并且因为这些基因在相同的途径中发挥功能的证据不存在。因此,通过用lenti-sgneo2/cre和lenti-sgsetd2/cre在kpt、kpt;cas9和klt;cas9小鼠中启动肿瘤来检验和确认setd2失活的背景依赖性。setd2失活增强了lkb1完整肺部的肿瘤生长,但很少(如果有的话)赋予lkb1缺陷肿瘤以生长优点(对于组织学分析,(kpt;cas9/klt;cas9中的sgsetd2)/(kpt;cas9/klt;cas9中的sgneo2)的p<0.05,对于tuba-seq分析,p<0.0001,图40和46)。该观察还得到了人肺部腺癌中lkb1/stk11和setd2改变的相互排斥性的很好支持(p<0.001,图40和46)。这些研究中的大多数基因表现出背景依赖性生长作用,仅在p53或lkb1存在或不存在的情况下驱动肿瘤生长(图40)。即使在全部三种情形下赋予优点的肿瘤抑制物改变(rb1和apc)也表现出肿瘤抑制的背景依赖性程度。这种广泛的背景依赖性在驱动子的全局调查中被忽略,其中驱动子相互作用被忽略或被认为是足够罕见和/或微弱的,以至于仅考虑边缘相关性是合理的。然而,我们的适合度测量总体上与人肺癌中的突变共存在模式相符,尽管这些数据的统计学分辨率有限(斯皮尔曼(spearman)r=0.50,p<0.05,图47)。此外,虽然肺癌在背景依赖性程度方面似乎不是唯一的(图47),并且这些发现表明在其他癌症类型中背景依赖性的直接测量是有保障的。肿瘤进化的这种崎岖景观具有若干含义。首先,为了理解基因功能,在多个遗传背景下研究推定的驱动子可以是重要的,因为调查中的大多数基因(11个中的8个)仅在一些背景下具有适应性(图40)。其次,更广泛的适合度谱分析是所期望的。这里的功效分析表明,使用tuba-seq与一百只小鼠的队列可以调查约500种中等强度的相互作用(图48)。较大的基因组筛选可以调查更多的推定驱动子、与其他致癌事件的相互作用、多个靶向相同的基因的sgrna或肿瘤抑制物改变的三联体。最后,这种广泛的背景依赖性表明,大多数驱动子改变很少会转移到固定,因为它们仅在特定遗传背景下有利。本文所述的体内组合肿瘤抑制物丧失的适合效应研究鉴定到出乎意料的遗传相互作用,这些相互作用已由传统方法以及由人肺部腺癌基因组学数据进行了验证。本文所述的具有条形码的和多重的基因组编辑方法可以容易地用于查询这些遗传相互作用的功能结果,包括它们对治疗响应、细胞信号传导和/或转移性进展的影响。实施例4:在hdr/sgrna/cre小鼠中通过药物筛选检测癌基因基因型-药物相互作用以与例如实施例2和图23a-d中描述的aav-krashdr/sgkras/cre小鼠相似的方法,产生包含在特定癌基因(例如hras、kras、pik3ca、pik3cb、egfr、pdgfr、vegfr2、her2、src、syk、abl、raf、myc或来自表1的任何基因)中具有多个不同的激活密码子突变的肿瘤的小鼠,并且在筛选过程中使用这些小鼠来鉴定癌基因基因型-药物相互作用,图49中说明了kras的情况。这是通过用与实施例2和图23a中描述的那些类似设计的cre腺病毒库来感染启动子-lsl-cas9(例如h11lsl-cas9)小鼠而实现的,该cre腺病毒携带:a)靶向靠近要引入激活突变处的特定目标癌基因的相关区域的sgrna;和b)hdr模板,其含有同源臂的、激活密码子突变(在每个构建体之间不同)、条形码序列(例如通过在激活突变下游的3、4、5、6、7、8、9或10个密码子内来自野生型序列的摆动碱基的变异而引入的),以及sgrna靶向位点处的任选pam突变,以防止再次裂解经hdr修复的基因组区域。在癌基因是kras的情况下,hdr模板含有例如kras中g12d、g12a、g12s、g12r、g12c、g12v的密码子突变,以及g13d外显子2突变。在一些实施方案中,在腺病毒库生成之后,改变库中每个密码子突变的组成(例如,在kras的情况下为g12d、g12a、g12s、g12r、g12c、g12v和/或g13d),使得施用给lsl-cas9小鼠时,携带单独密码子突变的每种病毒的剂量生成相同数量的肿瘤(例如在kras的情况下,通过使用与图23b中类似的信息生成)。然后全身性地或以组织特异性方式向小鼠施用cre腺病毒的多激活突变库(无论是考虑到不同突变的肿瘤形成效率而进行调节,还是未进行调节)(例如,气管内施用以诱导肺部肿瘤)。在用aav-cre病毒感染后,允许小鼠静止一段时间(例如12周)以允许肿瘤生长,然后用特定的化学治疗剂(例如,广谱烷化剂或微管蛋白靶向剂(诸如顺铂和紫杉醇),或靶向试剂(诸如mek抑制剂、erk抑制剂、mtor抑制剂和/或pi3k抑制剂))治疗数周时间(例如4周)。然后收获受感染的器官(例如在气管内施用病毒的情况下为肺部),从成批组织中分离基因组dna,并且如实施例2中那样进行癌基因和条形码的扩增和深度测序以测定组织中每种肿瘤的数量、大小和基因型。对相应的aav-cre感染的lsl-cas9小鼠进行类似分析,仅用媒介物代替化学治疗剂来治疗小鼠。使用如实施例2中所进行的类似生物信息学分析来测定经处理和未处理的动物之间,以及同一动物中源自不同激活癌基因突变的肿瘤之间(例如krasg12d、g12a、g12s、g12r、g12c、g12v和/或g13d)的肿瘤数量/大小的差异。具体而言,在用化学疗法治疗的同一动物内不同基因型肿瘤之间的肿瘤大小的比较允许以更高的精确性检测对药物的基因型特异性敏感性,因为数据不会因肿瘤启动速率或肿瘤生长速率的生物体间变异性而有偏。实施例5:用于条件性sgrna表达的慢病毒载体的设计为了实现对肿瘤启动事件和后续基因组改变的依次控制,设计了含cre以及flp调节的u6-sgrna元件的慢病毒载体。该载体旨在允许具有kraslsl-g12d等位基因的小鼠中cre/lox介导的肿瘤启动,同时实现通过诱导型flp活性对sgrna表达进行后续诱导。该载体中的关键设计特征是在sgrna阅读框上游的u6启动子内并入侧接两个杂交tata-frt位点的终止盒(例如seqidno:85′-gaagttcctattctctataaagtataggaacttc-3′)。这允许在u6启动子内并入终止盒,并且仅在flp介导的重组之后具有正确定位以指导转录的tata框。我们称这种设计为诱导型稳定sgrna表达;pinsane。因为在基因组整合后病毒载体由于位置效应和周围的染色质环境而变沉默,所以我们还靠近sgrna和u6启动子并入泛染色质开放元件(ucoe)。该ucoe源自人cbx3基因中的无甲基化cpg岛,并且已经证实以维持异源近端启动子的转录活性。该ucoe可以包括来自例如muller-kuller等人nucleicacidsres.2015年2月18日;43(3):1577–1592的任何ucoe(例如来自图s3的cbx或cbx3*)。示例性ucoe是下面的seqidno:9可以使用的ucoe的其他实例包括来自zhang等人molther.2010年9月;18(9):1640-9.doi:10.1038/mt.2010.132的那些。从pinsane载体中有效诱导sgrna需要可调节的flp活性。这可以通过并入在配体诱导型系统的控制下的flp(例如flp与在不存在配体的情况下阻断其活性的一个或多个结构域的蛋白质融合,或并入在配体诱导型启动子的控制下的flp基因)来实现。此类系统的一个实例是rosa26敲入等位基因,该等位基因含有与雌激素受体融合的密码子优化的flpo(r26flpoer(t2)),可以将其并入到我们的模型中以生成kraslslg12d/+;h11lsl-cas9;r26flpoer(t2)/lsl-tomato小鼠(ktc;flpoer)。该r26flpoer(t2)等位基因使他莫西芬(tam)诱导的核转位和flpoer的活性成为可能。这种等位基因已用于类似体内肺肿瘤模型中诱导flp活性,并在tam施用后在体内肺肿瘤中具有活性。配体诱导型系统的其他实例包括在图50中描绘了使用cre和flpoer重组酶的这种系统的实例,以及作为诱导剂的他莫西芬。图50a描绘了flp重组前后的示例性构建体的结构,而图50b描绘了靶向不同肿瘤抑制基因的sgrna构建体的示例性库。图50c描绘了当引入各种转基因背景的小鼠体内时,在有和无他莫西芬治疗的情况下,这种构建体的预期行为。实施例6:在双重sgrna小鼠中通过肿瘤分析检测成对肿瘤抑制物间的相互作用的策略如实施例1-3中那样构建小鼠,仅用病毒载体感染,所述病毒载体携带两个编码独特sgrna序列的u6-sgrna元件的,连同唯一性标识两个sgrna的组合的条形码序列(sgid)以及任选地,标识产生单独肿瘤(bc)的核酸分子的唯一分子标识符序列(umi)(图51a)。用需要筛选那样多的肿瘤抑制物的成对组合(例如,来自上表2的两种肿瘤抑制物的组合)来构建病毒载体。在一些实施方案中,将病毒构建体引入小鼠体内,该小鼠已经携带编码携带激活突变的癌基因的cre可激活的转基因(例如图51b的kt、kpt、klt小鼠,这些小鼠全部携带lsl-kras激活等位基因),从而允许在给定的癌基因背景下评估肿瘤抑制物的成对组合的作用。在一些实施方案中,将病毒构建体引入尚未携带癌基因突变的小鼠体内,从而允许在给定的癌基因背景下评估肿瘤抑制物的成对组合的作用。病毒构建体全身性地或以组织特异性方式施用(例如,对于肺部而言是在气管内施用),并且在肿瘤达到适于分析的大小(例如15周)后分离出所需组织或组织的组合。由该组织制备成批dna,并且如实施例1-3中那样扩增整合的病毒构建体的条形码区域并通过深度测序进行分析。使用如实施例2中所进行的类似生物信息学分析测定在有或无激活癌基因背景的情况下肿瘤抑制指导rna的不同成对组合之间肿瘤数量/大小的差异。虽然本文已经显示和描述了本发明的优选实施例,但是对于本领域技术人员显而易见的是,此类实施例仅以举例的方式提供。在不脱离本发明的情况下,现在本领域技术人员将想到许多改变、变化和取代。应该理解的是,本文所述的本发明的实施例的各种替代方案可用于实践本发明。其意图是以下权利要求限定本发明的范围,并且由此覆盖这些权利要求及其等同项范围内的方法和结构。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1