基于不同取代吡嗪受体单元的D-A共聚物给体材料及应用的制作方法
【技术领域】
本发明属于电化学材料技术领域,尤其涉及基于不同取代吡嗪受体单元的d-a共聚物给体材料及应用。
背景技术:
近些年来,基于富勒烯及非富勒烯受体的有机太阳能电池得到很大的发展,尤其是基于非富勒烯受体材料的有机聚合物发展的尤为迅速。基于d-a型共轭聚合物为给体非富勒烯材料为受体的有机太阳能电池的能量转化效率更是迅速超过了16%。非富勒烯受体材料发展的如此迅速,以至于能匹配的聚合物给体材料相对比较有限,发展比较缓慢。因此开发新型的聚合物给体材料具有很大的实际意义。
技术实现要素:
本发明的目的是提供基于不同取代吡嗪受体单元的d-a共聚物给体材料及应用,具有较强的光吸收能力、较好的电荷传输性能以及合适的电子能级。
本发明采用以下技术方案:基于不同取代吡嗪受体单元的d-a共聚物给体材料,该给体材料的通式如下:
其中:r选择h,f,cl,ome或coor’,r’选自c6~c20中的一种;r”选自h,f,cl或c6~c14中的一种;donors表示给体单元。
进一步地,该给体单元选自如下的一种:
其中:r1选自h,f或cl;r2选自c6~c14中的一种。
本发明还公开了基于不同取代吡嗪受体单元的d-a共聚物给体材料的制备方法,该制备方法如下:
步骤一、化合物a与化合物b通过stille偶联反应得到化合物c:
其中:r选择h,f,cl,ome或coor’,r’选自c6~c20;r”选自h,f,cl或c6~c14中的一种;
步骤二、将化合物c进行nbs溴代反应得到化合物d:
步骤三、将化合物d与donors聚合即得;所述donors为以下结构中的任意一种:
进一步地,该stille偶联反应条件为:溶剂为thf,催化剂为pd2(dba)3和三(邻甲基苯基)膦,催化剂加入量为化合物a摩尔量的5%~20%。优化的方案,化合物a和化合物b的摩尔比为1:2.5~3;在氮气保护下,80℃温度加热反应8~12小时。
进一步地,该nbs溴代反应条件为:溶剂thf,化合物c和nbs的摩尔比为1:3~4;避光,室温下反应48小时。
本发明还公开了上述基于不同取代吡嗪受体单元的d-a共聚物给体材料作为电子给体材料在有机太阳能电池中的应用。
本发明还公开了一种太阳能电池,选用上述的基于不同取代吡嗪受体单元的d-a共聚物给体材料作为电子供体。
本发明的有益效果是:1.合成的基于吡嗪单元的d-a共聚物结构新颖、简单,易于合成和制备。2.合成的基于吡嗪单元的d-a共聚物中的受体单元部分可溶于二氯甲烷、三氯甲烷、四氢呋喃等常见有机溶剂,易于溶液加工。3.合成的基于吡嗪单元的d-a共聚物有很宽的吸收光谱,利于吸收太阳光中的能量。4.合成的基于吡嗪单元的d-a共聚物有效的降低电池的humo能级,适合用于有机太阳能电池中的电子给体材料。5.合成的基于吡嗪单元的d-a共聚物作为电子给体材料有效地调节有机太阳能电池的开路电压和填充因子,在太阳能电池中展示出非常高的能量转换效率。
【附图说明】
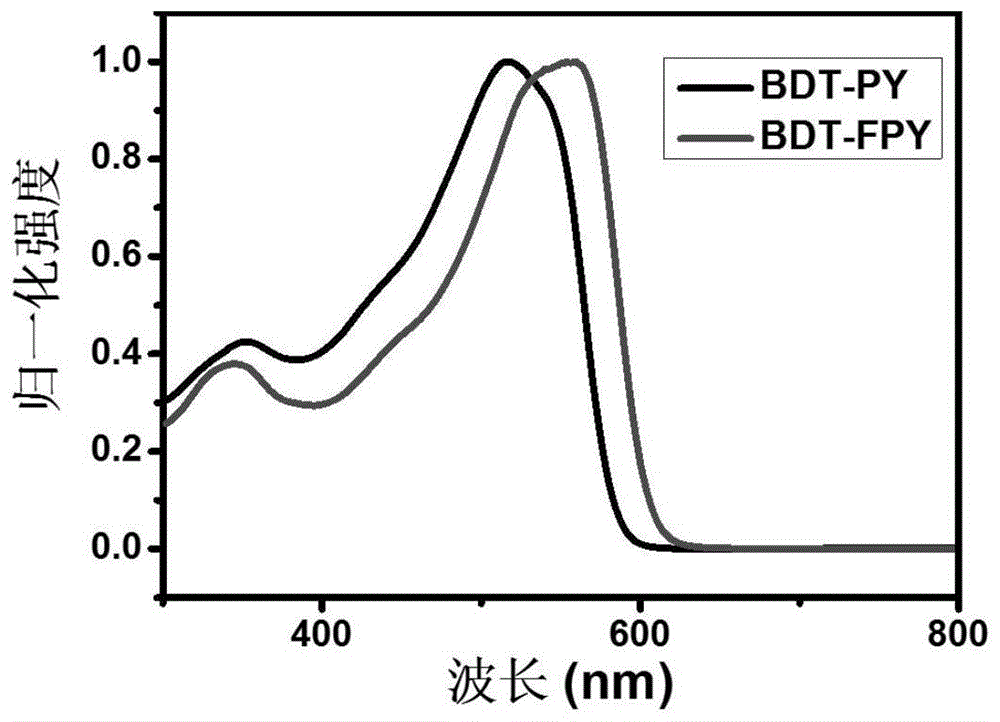
图1为本发明实施例1制备得到的bdt-py和实施例2制备得到的bdt-fpy的溶液中紫外-可见吸收光谱图。
图2为本发明实施例1制备得到的bdt-py和实施例2制备得到的bdt-fpy的循环伏安曲线。
图3为实施例1中,化合物c1的氢谱数据图。
图4为实施例1中,化合物c1的碳谱数据图。
图5为实施例1中,化合物d1的氢谱数据图。
图6为实施例1中,化合物d1的碳谱数据图。
图7为实施例2中,化合物c2的氢谱数据图。
图8为实施例2中,化合物c2的碳谱数据图。
图9为实施例2中,化合物c2的氟谱数据图。
图10为实施例2中,化合物d2的氢谱数据图。
图11为实施例2中,化合物d2的碳谱数据图。
图12为实施例2中,化合物d2的氟谱数据图。
图13为实施例1中,化合物d1的质谱数据图。
图14为实施例2中,化合物d2的质谱数据图。
图15为实施例3中,化合物c3的氢谱数据图。
图16为实施例4中,化合物c1的碳谱数据图。
图17为实施例4中,化合物d3的氢谱数据图。
图18为实施例4中,化合物d3的碳谱数据图。
图19为本发明实施例5制备得到的bdt-py和实施例6制备得到的bdt-fpy的薄膜状态下紫外-可见吸收光谱。
图20为本发明实施例5制备得到的bdt-py有机太阳能电池和实施例6制备得到的bdt-fpy有机太阳能电池的光电转化效率曲线。
图21为本发明实施例5制备得到的bdt-py和实施例6制备得到的bdt-fpy的有机太阳能电池的j-v曲线。
图22为本发明实施例5的giwaxs。
图23为本发明实施例6的giwaxs。
图24为本发明实施例5制备得到的bdt-py薄膜和实施例6制备得到的bdt-fpy薄膜的空穴迁移率数据图。
【具体实施方式】
下面结合附图和具体实施方式对本发明进行详细说明。
本发明实施例公开了基于不同取代吡嗪受体单元的d-a共聚物给体材料,该给体材料的通式如下:
其中:r选择h,f,cl,ome或coor’,r’选自c6~c20中的一种;r”选自h,f,cl或c6~c14中的一种;donors表示给体单元。r”可同时或者不同时选自同一取代基。
上述给体单元选自如下的一种:
其中:r1选自h,f或cl;r2选自c6~c14中的一种。r1可同时选自同一或者不同时选自同一取代基。同样,r2同时选自同一或者不同时选自同一取代基。
本发明公开了上述基于不同取代吡嗪受体单元的d-a共聚物给体材料在有机太阳能电池中的应用。
本发明公开了一种太阳能电池,选用上述的基于不同取代吡嗪受体单元的d-a共聚物给体材料作为电子供体。
上述基于不同取代吡嗪受体单元的d-a共聚物给体材料的制备方法包括以下步骤:
步骤一、化合物a与化合物b通过stille偶联反应得到化合物c:
其中:r选择h,f,cl,ome或coor’,r’选自c6~c20中的一种;r”选自h,f,cl或c6~c14中的一种。
上述的stille偶联反应条件为:溶剂为thf,催化剂为pd2(dba)3和三(邻甲基苯基)膦,催化剂加入量为化合物a摩尔量的5%~20%。优化的方案,化合物a和化合物b的摩尔比为1:2.5~3;在氮气保护下,80℃温度加热反应8~12小时。
步骤二、将化合物c进行nbs溴代反应得到化合物d:
上述的nbs溴代反应条件为:溶剂thf,化合物c和nbs的摩尔比为1:3~4;避光,室温下反应48小时;
步骤三、将化合物d与donors聚合得到新型的d-a共聚物给体材料,所述donors为以下结构中的任意一种:
上述的聚合反应条件为:溶剂为无水甲苯,催化剂为pd2(dba)3和三(邻甲基苯基)膦。化合物d和供体材料donors的摩尔比为1:1~2,氩气保护下110℃反应。
实施例1:
bdt-py的合成路线如下:
(1)化合物a1与化合物b1通过stille偶联反应得到化合物c1:
具体为:称取化合物a1(1.5g,6.3mmol),化合物b1(6.8g,14.0mmol),pd2(dba)3(0.6g,0.6mmol),三(邻甲基苯基)膦(0.4g,1.3mmol)于250ml反应管中,在氮气保护下,加入thf50ml,于80℃反应过夜。反应结束冷却至室温,饱和氟化钾溶液洗三次,二氯甲烷萃取,收集有机相用无水na2so4进行干燥,旋蒸除去大部分有机液体后,使用硅胶柱色谱进行纯化(pe/dcm=1/1,v/v),收集目标化合物。
产物为黄色固体(1.5g),即为化合物c1,如图3、图4所示,对化合物c1的结构进行了确证,1hnmr(600mhz,cdcl3)δ[ppm]8.81(s,2h),7.46(s,2h),7.03(s,2h),2.58-2.57(d,j=6hz,4h),1.62-1.59(m,2h),1.33-1.29(m,16h),0.91-0.89(m,12h);13cnmr(cdcl3,100mhz):δ[ppm]:145.50,143.11,140.25,138.89,126.58,123.80,39.81,34.09,31.95,28.38,25.11,22.56,13.66,10.35。
(2)将化合物c1与nbs进行溴代反应得到化合物d1:
称取化合物c1(1.5g,3.2mmol)和nbs(2.4g,13.4mmol)于250ml圆底烧瓶中,加入thf50ml,避光,室温下反应24小时。反应结束后,抽滤除去不溶性杂质,旋蒸除去大部分液体,用硅胶柱色谱进行纯化(pe/dcm=1/1,v/v),收集目标化合物。
产物为黄色固体1.3g,即为化合物d1,如图5,图6和图13所示,对化合物d1结构进行确证,1hnmr(600mhz,cdcl3)δ[ppm]8.72(s,2h),7.30(s,2h),2.53-2.52(d,j=6hz,4h),1.68-1.53(m,2h),1.37-1.26(m,16h),0.92-0.88(m,12h);13cnmr(cdcl3,100mhz):δ[ppm]:144.35,141.97,139.49,137.85,125.43,112.93,38.94,32.92,31.42,27.74,26.64,22.03,13.11,9.81。化合物d1的质谱数据为,c28h38br2n2s2的maldi-tofms(m/z):626.08。
(3)化合物d1通过聚合反应反应得到bdt-py:
向配备有搅拌棒的25ml反应管中添加d1(62.6mg,0.1mmol),c4,c6bdt单体(101.7mg,0.1mmol),pd2(dba)3(1.37mg,0.0015mmol),三(邻甲苯基)磷(3.65mg,0.012mmol)和无水甲苯(4.0ml)。用氩气吹扫反应管,并在氩气流下密封。将该管加热至110℃后反应24小时。冷却至室温后,在剧烈搅拌下将反应溶液滴加到80ml甲醇中,并搅拌1h。过滤沉淀物,并通过硅胶柱(chcl3)纯化。得到呈紫红色固体的产物即bdt-py(108.8mg)。
bdt-py的溶液紫外-可见吸收光谱如图1所示,bdt-fpy的氯仿溶液在400-630nm区域表现出较强的吸收性,有很宽的吸收光谱,利于吸收太阳光中的能量。循环伏安曲线如2所示,通过循环伏安曲线测定的氧化还原电位计算bdt-py的homo和lumo能级,其homo能级为-5.44ev,lumo能级为-3.56ev。图19为本发明实施例1制备得到的bdt-py的薄膜状态下紫外-可见吸收光谱,bdt-py的最大吸收从溶液中的518nm迁移到572nm,这表明共聚物可以在薄膜中形成紧密的分子团。
实施例2:
bdt-fpy的合成路线如下:
(1)3,6-二溴-吡嗪-2-胺和氟硼酸制备化合物a2:
称取3,6-二溴-吡嗪-2-胺(10.0g,39.5mmol)和nano2(10.8g,156.5mmol)于500ml圆底烧瓶中,在0℃下用恒压滴液漏斗滴加氟硼酸100ml,室温下反应12小时。反应结束后,水洗三次,再用二氯甲烷萃取,收集有机相,无水mg2so4干燥,旋蒸除去大部分溶剂,使用硅胶柱色谱进行纯化(pe/dcm=10/1,v/v),收集目标化合物。
产物为白色固体(5.4g),即为化合物a2。
(2)化合物a2与化合物b1通过stille偶联反应得到化合物c2:
称取化合物a2(1.5g,5.9mmol),化合物b1(5.0mg,13.4mol),pd2(dba)3(0.5g,0.5mmol),三(邻甲基苯基)膦(0.4g,1.3mmol)于250ml反应管中,在氮气保护下,加入thf50ml,于80℃反应过夜。反应结束冷却至室温,饱和氟化钾溶液洗三次,二氯甲烷萃取,收集有机相用无水na2so4进行干燥,旋蒸除去大部分有机液体后,使用硅胶柱色谱进行纯化(pe/dcm=1/1,v/v),收集目标化合物。
产物为黄色固体1.7g,即为化合物c2,如图7,图8,图9所示,对化合物结构进行确证,1hnmr(600mhz,cdcl3)δ[ppm]8.68-8.67(d,j=6hz,1h),7.64(s,1h),7.51(s,1h),7.09(s,1h),7.05(s,1h)2.59-2.56(dd,j=6hz,12hz,4h),1.62-1.59(m,2h),1.34-1.29(m,16h),0.92-0.89(m,12h);13cnmr(cdcl3,100mhz):δ[ppm]:155.46,153.75,143.77,143.41,143.36,138.81,137.58,137.52,136.34,134.78,134.60,130.65,130.58,128.39,125.29,125.03,40.32,40.31,34.64,34.55,32.46,28.88,25.64,25.62,23.09,14.18,10.86。
(3)将化合物c2与nbs进行溴代反应得到化合物d2:
称取化合物c2(1.7g,3.5mmol)和nbs(2.5g,14.0mmol)于250ml圆底烧瓶中,加入thf50ml,避光,室温下反应24小时。反应结束后,抽滤除去不溶性杂质,旋蒸除去大部分液体,用硅胶柱色谱进行纯化(pe/dcm=1/1,v/v),收集目标化合物。
产物为黄色固体1.5g,即为化合物d2,如图10,图11,图12和图14所示,对化合物d2的结构进行确证,1hnmr(600mhz,cdcl3)δ[ppm]8.60-8.59(d,j=6hz,1h),7.47(s,1h),7.36(s,1h),2.54-2.51(m,4h),1.67-1.65(m,2h),1.35-1.30(m,16h),0.92-0.90(m,12h);13cnmr(cdcl3,100mhz):δ[ppm]:155.24,153.53,143.27,142.64,138.42,137.35,137.29,135.92,134.23,134.05,130.25,127.84,115.43,114.74,39.96,33.98,32.42,28.76,25.67,23.05,14.13,10.83。化合物d2的质谱数据为,c28h38br2n2s2f的maldi-tofms(m/z):645.08。
(4)化合物d2通过聚合反应得到bdt-fpy:
向配备有搅拌棒的25ml反应管中添加d2(64.4mg,0.1mmol),c4,c6bdt单体(101.7mg,0.1mmol),pd2(dba)3(1.37mg,0.0015mmol),三(邻甲苯基)磷(3.65mg,0.012mmol)和无水甲苯(4.0ml)。用氩气吹扫反应管,并在氩气流下密封。将管加热至110℃持续1小时。冷却至室温后,在剧烈搅拌下将反应溶液滴加到80ml甲醇中,并搅拌1h。过滤沉淀物,并通过硅胶柱(chcl3)纯化,得到深红色固体状的产物聚合物bdt-fpy(104.6mg)。
bdt-fpy的紫外-可见吸收光谱如图1所示,bdt-fpy的氯仿溶液在400-630nm区域表现出较强的吸收性,有很宽的吸收光谱,利于吸收太阳光中的能量。循环伏安曲线如图2所示,通过循环伏安曲线测定的氧化还原电位计算bdt-fpy的homo和lumo能级,其homo能级为-5.48ev,lumo能级为-3.70ev。图19为本发明实施例2制备得到的bdt-fpy的薄膜状态下紫外-可见吸收光谱,bdt-fpy的最大吸收从溶液中的554nm迁移到591nm,这表明共聚物可以在薄膜中形成紧密的分子团。
实施例3:
bdt-py20的合成路线如下:太阳能光伏器件的制备及性能测试:
(1)3,6-二溴吡嗪-2-甲酯制备化合物a3:
称取3,6-二溴吡嗪-2-甲酯(5.0g,16.9mmol)和一水合氢氧化锂(1.5g,35.7mmol)于250ml圆底烧瓶中,加入thf:甲醇:水=36ml:9ml:9ml,冰浴反应40分钟。反应结束后,加入1n的盐酸酸化至酸性,用二氯甲烷萃取三次,收集有机相,旋蒸除去大部分液体,得淡黄色油状液体5.0g,将所得油状液体(5.0g,17.7mmol),2-辛基十二醇(15.3g,51.2mmol),dcc(4.4g,21.3mmol),dmap(0.6g,4.9mmol)于250ml圆底烧瓶中,在氮气保护下加入二氯甲烷25ml,室温下反应48小时。反应结束后,抽滤除去不溶性杂质,收集滤液,旋蒸除去大部分有机液体后,使用硅胶柱色谱进行纯化(pe/ea=50/1,v/v),收集目标化合物。产物为黄色油状液体(5.2g)即为化合物a3。
(2)化合物a3与化合物b2通过stille偶联反应得到化合物c3:
称取化合物a3(3.4g,6.0mmol),化合物b2(9.1g,24.4mmol),pd2(dba)3(0.6g,0.6mmol),三(邻甲基苯基)膦(0.7g,2.3mmol)于250ml反应管中,在氮气保护下,加入thf50ml,于80℃反应过夜。反应结束冷却至室温,饱和氟化钾溶液洗三次,二氯甲烷萃取,收集有机相用无水na2so4进行干燥,旋蒸除去大部分有机液体后,使用硅胶柱色谱进行纯化(pe/dcm=5/1,v/v),收集目标化合物。
产物为黄色固体(1.8g),即为化合物c3,如图15,图16所示,对化合物结构进行确证,1hnmr(400mhz,cdcl3)δ[ppm]8.93(s,1h),7.73-7.72(dd,j=1.2hz,4hz,1h),7.50-7.46(m,3h),7.15-7.13(dd,j=4hz,8hz,1h),7.11-7.09(dd,j=4hz,8hz,1h),4.32-4.31(d,j=4hz,2h),1.73(s,1h),1.29-1.25(m,32h),0.90-0.86(m,6h);13cnmr(cdcl3,100mhz):δ[ppm]:166.74,144.88,142.68,142.27,140.31,140.09,139.98,129.39,129.31,128.34,128.20,127.47,126.26,69.20,37.17,31.87,30.90,29.88,29.63,29.5929.55,29.50,29.30,26.62,22.63,14.07。
(3)将化合物c3与nbs进行溴代反应得到化合物d3:
称取化合物c3(1.5g,2.6mmol),nbs(2.0g,11.6mmol)于250ml圆底烧瓶中。加入thf30ml,室温下避光反应48h。反应结束后,抽滤除去不溶性杂质,旋蒸除去大部分液体,用硅胶柱色谱进行纯化(pe/dcm=5/1,v/v),收集目标化合物。
产物为黄色固体(1.8g),即为化合物d3。如图17,图18所示,对化合物d3的结构进行确证,1hnmr(400mhz,cdcl3)δ[ppm]8.81(s,1h),7.44-7.43(d,j=4hz,1h),7.23-7.22(d,j=4hz,1h),7.10-7.09(d,j=4hz,1h),7.04-7.03(d,j=4hz,1h),4.32-4.30(d,j=8hz,2h),1.75(s,1h),1.30-1.26(m,32h),0.89-0.86(m,6h);13cnmr(cdcl3,100mhz):δ[ppm]:165.28,143.05,141.03,140.83,140.66,140.56,138.40,130.35,130.23,126.93,125.30,116.73,116.52,68.41,36.27,30.91,30.05,28.97,28.69,28.64,28.59,28.35,25.70,21.68,13.13。
(4)化合物d3通过聚合反应反应得到bdt-py20:
向配备有搅拌棒的25ml反应管中添加d3(72.7mg,0.1mmol),c2,c4bdt单体(90.6mg,0.1mmol),pd2(dba)3(1.38mg,0.0015mmol),三(邻甲苯基)磷(3.63mg,0.012mmol)和无水甲苯(4.0ml)。用氩气吹扫反应管,并在氩气流下密封。将管加热至110℃持续45分钟。冷却至室温后,在剧烈搅拌下将反应溶液滴加到80ml甲醇中,并搅拌1h。过滤沉淀物,并通过硅胶柱(chcl3)纯化,得到深红色固体状的产物聚合物(109.5mg)。
实施例4:
化合物d3通过聚合反应反应得到fbdt-py20:
向配备有搅拌棒的25ml反应管中添加d3(72.5mg,0.1mmol),c2,c4fbdt单体(94.4mg,0.1mmol),pd2(dba)3(1.36mg,0.0015mmol),三(邻甲苯基)磷(3.66mg,0.012mmol)和无水甲苯(4.0ml)。用氩气吹扫反应管,并在氩气流下密封。将管加热至110℃持续45分钟。冷却至室温后,在剧烈搅拌下将反应溶液滴加到80ml甲醇中,并搅拌1h。过滤沉淀物,并通过硅胶柱(chcl3)纯化,得到深红色固体状的产物聚合物(113.6mg)。
实施例5
以实施例1中的化合物bdt-py为供体材料制备有机太阳能电池。
器件制备:以ito/pedot:pss/bdt-py:meic/zracac/al的常规装置构造来制造太阳能电池。首先用去污剂擦洗氧化铟锡(ito)玻璃基板,然后依次用去离子水,丙酮和异丙醇超声处理,然后在烤箱中干燥过夜。使用前,将玻璃基板用uv-臭氧处理30分钟。将聚乙撑二氧噻吩:聚苯乙烯磺酸盐(pedot:pss重量比为1:1)(heraeuscleviospvpai4083)以4000rpm的速度旋铸到ito基板上30s,然后在150℃干燥15分钟。将供体:受体混合物(重量比为1:1)溶解在氯仿中,所有混合物的混合物溶液总浓度为16mgml-1,选择dio作为体积比为0.25%的添加剂,并在室温下搅拌过夜。将共混溶液以3000rpm旋转浇铸30s。涂覆后,将活性层在100℃的热板上退火3分钟。在活性层上涂覆zracac薄层,然后在活性层上真空5×10-5pa蒸镀厚度100nm左右的金属铝作为光伏器件的阴极。
器件测试条件:通过brukerdektakxt测针轮廓仪测得的最佳活性层厚度约为105nm。使用keithley2400离子源测量仪,在空气中,am1.5g(100mwcm-2)下,使用newport太阳模拟器,测量了所有封装器件的电流密度-电压(j-v)曲线。使用标准的si二极管校准光强度。标准的si二极管带kg5滤光片,购自pvmeasurement,以使光谱失配达到统一。使用光学显微镜(olympusbx51)定义设备面积(5.9mm2)。使用配备了标准si二极管的enlitechqe-seqe系统测量eqe。单色光是由newport300w光源产生的。
图20为本发明实施例5制备得到的bdt-py有机太阳能电池的光电转化效率曲线。其外部量子效率(eqe)光谱在300-600nm的波长范围内表现出强的,宽的光响应,从eqe曲线计算得出的基于bdt-py器件的jsc值分别为15.54macm-2,这与从j-v曲线得出的值基本一致。
如图21所示为本发明实施例5制备得到的bdt-py的有机太阳能电池的j-v曲线;测得实施例5器件的短路电流jsc为15.1macm-2,开路电压voc为0.95v,填充因子ff为68.5%,能量转换效率pce为10.2%。
如图22所示,进行了giwaxs实验以研究共混膜的分子堆积和结晶。基于bdt-py:meic的混合膜表现出显着的面对堆叠模式,有利于电荷传输和电荷提取。
如图24所示,通过sclc技术测量了空穴(μh)迁移率。基于bdt-py的设备显示出高的空穴迁移率μh=3.23×10-4cm2v-1s-1这有利于电子传输。
本发明实施例5的器件中所用的受体的结构如下:
实施例6
以实施例2中的化合物bdt-fpy为供体材料制备有机太阳能电池
器件制备:以ito/pedot:pss/bdt-fpy:meic/zracac/al的常规装置构造来制造太阳能电池。首先用去污剂擦洗氧化铟锡(ito)玻璃基板,然后依次用去离子水,丙酮和异丙醇超声处理,然后在烤箱中干燥过夜。使用前,将玻璃基板用uv-臭氧处理30分钟。将聚乙撑二氧噻吩:聚苯乙烯磺酸盐(pedot:pss重量比为1:1)(heraeuscleviospvpai4083)以4000rpm的速度旋铸到ito基板上30s,然后150℃干燥15分钟。将供体:受体混合物(重量比为1:1)溶解在氯仿中(所有混合物的混合物溶液总浓度为16mgml-1,选择dio作为体积比为0.25%的添加剂),并在室温下搅拌过夜。将共混溶液以3000rpm旋转浇铸30s。涂覆后,将活性层在100℃的热板上退火3分钟。在活性层上涂覆zracac薄层,然后在活性层上真空(5×10-5pa)蒸镀厚度100nm左右的金属铝作为光伏器件的阴极。
器件测试条件:通过brukerdektakxt测针轮廓仪测得的最佳活性层厚度约为105nm。使用keithley2400离子源测量仪,在空气中,am1.5g(100mwcm-2)下,使用newport太阳模拟器,测量了所有封装器件的电流密度-电压(j-v)曲线。使用标准的si二极管,校准光强度。标准的si二极管带kg5滤光片,购自pvmeasurement以使光谱失配达到统一。使用光学显微镜(olympusbx51)定义设备面积(5.9mm2)。使用配备了标准si二极管的enlitechqe-seqe系统测量eqe。单色光是由newport300w光源产生的。
图20为本发明实施例5制备得到的bdt-fpy有机太阳能电池的光电转化效率曲线。其外部量子效率(eqe)光谱在300-600nm的波长范围内表现出强的,宽的光响应,从eqe曲线计算得出的基于bdt-fpy器件的jsc值为17.29macm-2,这与从j-v曲线得出的值基本一致。
图21为本发明实施例6制备得到的有机太阳能电池的j-v曲线测得,实施例6器件的短路电流jsc为17.0macm-2,开路电压voc为0.96v,填充因子ff为73.2%,能量转换效率pce为12.3%。
如图23示,进行了giwaxs实验以研究共混膜的分子堆积和结晶。基于bdt-fpy:meic的混合膜表现出显着的面对堆叠模式,有利于电荷传输和电荷提取。
如图24所示,通过sclc技术测量了空穴迁移率μh。基于bdt-fpy的设备显示出高的空穴迁移率μh=3.42×10-4cm2v-1s-1这有利于电子传输。
本发明实施例6的器件中所用的受体的结构如下:
氟、氯原子修饰的基于取代吡嗪受体单元的共轭聚合物给体材料可以降低有机太阳能电池的能级,改善材料的结晶性能,进而调节有机太阳能电池的开路电压和填充因子,从而改善有机太阳能电池的效率。也能为非富勒材料的开发提供更多的给体聚合物材料的选择。促进有机太阳能电池的向前发展。
本发明创新性的基于吡嗪作为受体单元,构建一种新型的d-a共聚物给体材料,本发明的聚合物给体材料可以吸收300-700nm左右的吸光范围,应用于有机太阳能电池可以有效的降低电池的humo能级,提高聚合物的结晶性能进而提高聚合物的能量转化效率。
- 还没有人留言评论。精彩留言会获得点赞!