一种布洛芬杂质I的环保制备方法与流程
本发明属于药物杂质标准品的制备
技术领域:
,具体涉及一种布洛芬杂质i的环保制备方法。
背景技术:
:布洛芬(ibuprofen),化学名为2-(4-异丁基苯基)丙酸,中文别名拔怒风,是一种临床上广泛使用的非甾体抗炎药,属于芳基烷酸类药物。布洛芬可作为阿司匹林的替代品,具有更强的解热、消炎和镇痛作用,主要用于感冒、风湿和类风湿关节炎等疾病中炎症和疼痛的控制,而副作用却比阿司匹林小很多。欧洲药典、英国药典、美国药典、药典、中国药典均收载了布洛芬这一品种,对其有关物质的测定也做了详细的阐述。布洛芬原料药中已报道的杂质有18种,包括,杂质a、b、c、d、e、f、g、h、i、j、k、l、m、n、o、p、q和r。杂质对原料药的质量控制十分重要,许多国家在药品质量研究指导中对药品杂质研究提出明确的技术要求,在进行质量研究时要得到其杂质标准品。目前,关于布洛芬杂质i的制备方法未见报道,尤其是高纯度布洛芬杂质i的制备方法。技术实现要素:本发明的一个目的在于提供一种具有操作简单、制备周期短、副产物少、易于纯化、产率高、环保的优点,制备得到的布洛芬杂质i的纯度高,无明显杂质点的布洛芬杂质i的环保制备方法。本发明为实现上述目的所采取的技术方案为:一种布洛芬杂质i的环保制备方法,包括以下步骤:向含布洛芬杂质h和zn的etoh溶液中加入nh4cl和hcl,在50-90℃搅拌反应10-15h,过滤并蒸发溶剂,柱层析分离纯化,得到布洛芬杂质i。本发明方法具有操作简单、制备周期短、副产物少、易于纯化、产率高、环保的优点,制备得到的布洛芬杂质i的纯度高,无明显杂质点,符合杂质对照品要求,可作为布洛芬杂质i标准品,应用于布洛芬杂质i的定性、定量研究和检测,对布洛芬及其相关制剂的质量控制具有一定的意义。优选的,布洛芬杂质h、zn和nh4cl的摩尔比为1:2:2.5-3.5。优选的,布洛芬杂质h和hcl的用量比为1:1.5-2.0(g/ml)优选的,柱层析分离纯化用洗脱剂为pe:ea=10-30:1。优选的,洛芬杂质i的产率至少为85.00%。优选的,洛芬杂质h通过如下方法制备:步骤1:式(i)所示化合物与式(ii)所示化合物在naoc2h5的催化作用下进行反应制备得式(iii)所示化合物;步骤2:式(iii)所示化合物在ch3mgbr的存在下反应制备得到布洛芬杂质h。布洛芬杂质h的制备方法具有操作简单、制备周期短、副产物少、易于纯化、产率高、环保的优点,制备得到的布洛芬杂质h的纯度高,无明显杂质点。本发明的又一目的,在于提供一种布洛芬杂质i,通过上述环保制备方法制备得到。优选的,洛芬杂质i的纯度至少为99.00%。本发明的又一目的,在于提供一种布洛芬杂质i的检测方法,所用标准品为权利要求7或8所述的布洛芬杂质i。本发明布洛芬杂质i具有较高的纯度,能够提高检测方法的准确度。优选的,检测方法的准确度好,布洛芬杂质i的平均回收率至少为99.90%,rsd%≤5%。与现有技术相比,本发明的有益效果为:本发明方法具有操作简单、制备周期短、副产物少、易于纯化、产率高、环保的优点,制备得到的布洛芬杂质i的纯度高,无明显杂质点,符合杂质对照品要求,可作为布洛芬杂质i标准品,应用于布洛芬杂质i的定性、定量研究和检测,对布洛芬及其相关制剂的质量控制具有一定的意义。本发明布洛芬杂质i具有较高的纯度,作为布洛芬杂质i标准品进行检测时,能够提高检测方法的准确度。本发明采用了上述技术方案提供一种布洛芬杂质i的环保制备方法,弥补了现有技术的不足,设计合理,操作方便。附图说明图1是本发明实施例1中布洛芬杂质h的ms谱图;图2是本发明实施例1中布洛芬杂质h的1hnmr谱图;图3是本发明实施例1中布洛芬杂质h的hplc谱图图4是本发明实施例1中布洛芬杂质i的ms谱图;图5是本发明实施例1中布洛芬杂质i的1hnmr谱图;图6是本发明实施例1中布洛芬杂质i的hplc谱图。具体实施方式本发明允许各种修改及变形,其特定实施例进行了举例,下面进行详细说明。但并非要把本发明限定于公开的特别形态之意,相反,本发明包括与由权利要求项所定义的本发明思想一致的所有修改、均等及替代。这些实施例只用于更具体地说明本发明,根据本发明的要旨,本发明的范围并非限定于这些实施例,这是所属
技术领域:
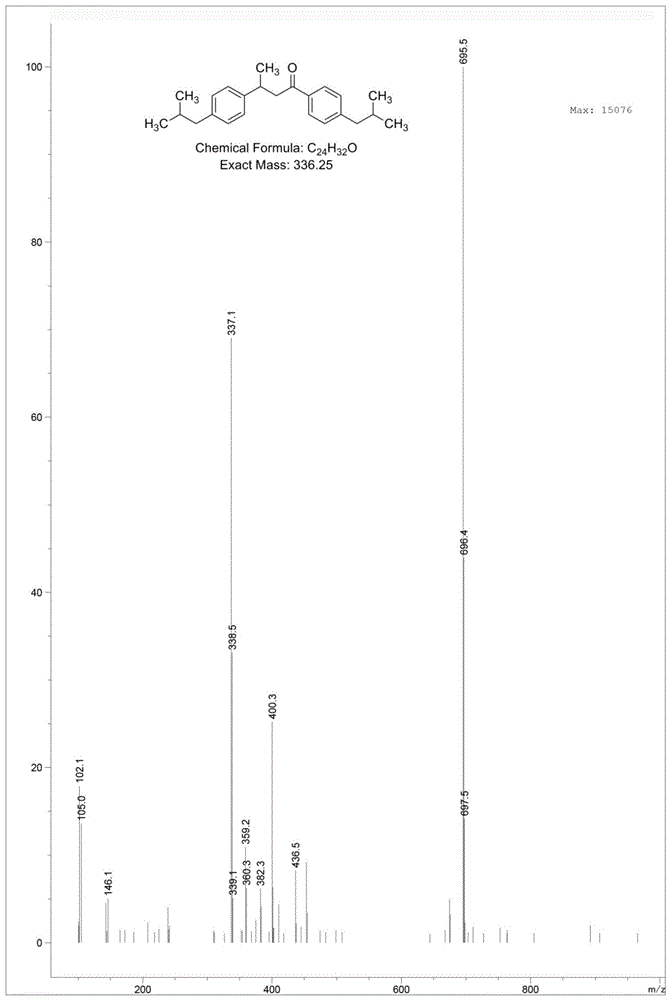
的技术人员不言而喻的。本发明一实施方式提供了一种布洛芬杂质i的环保制备方法,合成路径为:具体包括以下步骤:向含布洛芬杂质h和zn的etoh溶液中加入nh4cl和hcl,布洛芬杂质h、zn和nh4cl的摩尔比为1:2:2.5-3.5,布洛芬杂质h和hcl的用量比为1:1.5-2.0(g/ml),然后在50-90℃搅拌反应10-15h,过滤并蒸发溶剂,柱层析分离纯化,洗脱剂为pe:ea=10-30:1,得到布洛芬杂质i。本实施方法提供的制备方法具有操作简单、制备周期短、副产物少、易于纯化、产率高、环保的优点,洛芬杂质i的产率至少为85.00%,制备得到的布洛芬杂质i的纯度高,无明显杂质点,符合杂质对照品要求,可作为布洛芬杂质i标准品,应用于布洛芬杂质i的定性、定量研究和检测,对布洛芬及其相关制剂的质量控制具有一定的意义。于本发明一实施方式中,洛芬杂质h通过如下方法制备:步骤1:在60-80℃下向式(i)所示化合物与式(ii)所示化合物的etoh溶液中分批添加naoc2h5,式(i)所示化合物、式(ii)所示化合物和naoc2h5的摩尔比为1:1:1.5-2.3,共搅拌反应10-15h,将反应液旋蒸发除去etoh,再将得到的固体溶解,经柱层析分离纯化,洗脱剂为pe:ea=5-15:1,得到式(iii)所示化合物;步骤2:将式(iii)所示化合物溶于无水thf中,在-10至-30℃下,向其中缓慢加入浓度为1mol/l的ch3mgbr试剂,式(iii)所示化合物和ch3mgbr的摩尔比为1:1,在该温度下搅拌反应40-80min,将反应液旋蒸发除去thf,并通过柱层析分离纯化,洗脱剂为pe:ea=40-60:1,得到布洛芬杂质h。布洛芬杂质h的制备方法具有操作简单、制备周期短、副产物少、易于纯化、产率高、环保的优点,洛芬杂质h的产率至少为35.00%,制备得到的布洛芬杂质h的纯度至少为99.00%,无明显杂质点,从而提高布洛芬杂质i的得率和纯度。本发明一实施方式还提供了一种布洛芬杂质i,通过上述环保制备方法制备得到。于本发明一实施方式中,洛芬杂质i的纯度至少为99.00%。本发明一实施方式还提供了一种布洛芬杂质i的检测方法,采用hplc法,具体包括如下步骤:(1)供试品溶液的配制:称取布洛芬原料适量,精密称定,加50%乙腈溶液溶解并定量稀释制成每1ml含布洛芬为2mg的溶液,作为供试品溶液;(2)对照品溶液的配制:取上述布洛芬杂质i适量,精密称定,加50%乙腈溶液溶解并定量稀释制成每1ml含布洛芬杂质i为2.0μg的溶液,作为对照品溶液;所用布洛芬杂质i具有较高的纯度,能够提高检测方法的准确度;(3)样品检测:精密量取上述供试品溶液和对照品溶液各50μl,注入液相色谱仪,记录色谱图;其中,色谱条件如下:色谱柱:agilentec-c184.6×150mm2.7μm;流速:1.0ml/min;检测波长:254nm;进样量:5μl;柱温:25℃;流动相:a:20%乙腈;b:0.1%磷酸溶液。于本发明一实施方式中,检测方法的准确度好,布洛芬杂质i的平均回收率至少为99.90%,rsd%≤5%。以下通过实施例来进一步阐明本发明。但是应该理解,实施例只是举例说明的目的,并不意欲限制本发明的范围和精神。实施例1:一种布洛芬杂质i的合成路线为:一种布洛芬杂质i的环保制备方法,包括以下步骤:1)布洛芬杂质h的制备,具体步骤为:步骤1:将0.5g式(i)所示化合物与0.543g式(ii)所示化合物混合溶于etoh中,然后在60-80℃下将0.42gnaoc2h5分4批添加至其中,共搅拌反应12h,将反应液旋蒸发除去etoh,再将得到的固体溶解,经柱层析分离纯化,洗脱剂为pe:ea=10:1,得到式(iii)所示化合物;步骤2:将0.5g式(iii)所示化合物溶于无水thf中,在-20℃下,向其中缓慢加入1.56ml浓度为1mol/l的ch3mgbr试剂,式(iii)所示化合物和ch3mgbr的摩尔比为1:1,在该温度下搅拌反应60min,将反应液旋蒸发除去thf,并通过柱层析分离纯化,洗脱剂为pe:ea=50:1,得到200mg布洛芬杂质h,布洛芬杂质h的产率为38.10%;2)布洛芬杂质i的制备,向含0.3g布洛芬杂质h和0.116gzn的etoh溶液中加入0.143gnh4cl和0.5mlhcl,在70℃搅拌反应12h,反应结果后将反应液旋蒸发除去etoh,再将得到的固体溶解,柱层析分离纯化,洗脱剂为pe:ea=20:1,得到254mg布洛芬杂质i,布洛芬杂质i的产率为86.00%。布洛芬杂质h的结构式为:布洛芬杂质h的分子式:c24h32o;布洛芬杂质h的分子量:336.51。采用质谱仪对合成产物-布洛芬杂质h进行分析,其的ms谱图如图1所示,在质谱图中,m/z=337.1的峰为布洛芬杂质h的分子离子峰,与布洛芬杂质h的分子量吻合。同时谱图上未显示反应原料-式(iii)所示化合物的色谱及质谱峰,说明其转化率已达100%。采用核磁共振仪对合成产物-布洛芬杂质h进行分析,其的1hnmr谱图如图2所示,1hnmr(400mhz,dmso-d6):δ7.85-7.87(d,2h,ar-h),7.26-7.28(d,2h,ar-h),7.18-7.20(d,2h,ar-h),7.03-7.05(d,2h,ar-h),3.19-3.33(m,3h,ch,ch2),2.50-2.54(d,2h,ch2),2.37-2.39(d,2h,ch2),1.74-1.84(m,2h,ch),1.21-1.22(d,3h,ch3),0.83-0.86(d,12h,ch3)。合成产物-布洛芬杂质h纯度分析采用hplc检测方法,hplc检测方法的条件如下:色谱柱:agilentec-c184.6×150mm2.7μm;流速:0.3ml/min;检测波长:sig=254nm;进样量:5μl;柱温:25℃;流动相:a:95%乙腈;b:0.1%甲酸溶液;按表1进行梯度洗脱程序;时间:18min。图3是合成产物-布洛芬杂质h的hplc谱图,hplc谱图中各峰分析结果如表2,可以看出,合成产物-布洛芬杂质h纯度为99.64%。表1流动相比例time(min)a(%)b(%)0.0095518.00955表2hplc谱图中各峰分析结果peak#rettime(min)areaheightarea%10.7536.476762.079810.040520.93613.373983.097450.083631.29418.226274.026250.114044.36318.831002.075880.117854.9261.59354e41673.8685399.6442totals1.59923e41685.14792100.0000布洛芬杂质i的结构式为:布洛芬杂质i的分子式:c24h34;布洛芬杂质i的分子量:322.24。采用质谱仪对合成产物-布洛芬杂质i进行分析,其的ms谱图如图4所示,在质谱图中,m/z=321.3的峰为布洛芬杂质i的分子离子峰,与布洛芬杂质i的分子量吻合;m/z=336.6的峰为布洛芬杂质h的分子离子峰,与布洛芬杂质i的分子量吻合。采用核磁共振仪对合成产物-布洛芬杂质i进行分析,其的1hnmr谱图如图5所示,1hnmr(400mhz,dmso-d6):δ7.09-7.13(d,4h,ar-h),7.03-7.07(d,4h,ar-h),2.50-2.51(m,1h,ch),2.38-2.40(d,6h,ch2),1.79-1.82(m,4h,ch,ch2),1.19-1.21(d,3h,ch3),0.83-0.87(d,12h,ch3)。合成产物-布洛芬杂质i纯度分析采用hplc检测方法,hplc检测方法的条件如下:色谱柱:agilentec-c184.6×150mm2.7μm;流速:0.3ml/min;检测波长:sig=254nm;进样量:5μl;柱温:25℃;流动相:a:20%乙腈;b:0.1%磷酸溶液;按表3进行梯度洗脱程序;时间:42min。图6是合成产物-布洛芬杂质i的hplc谱图,hplc谱图中各峰分析结果如表4,可以看出,合成产物-布洛芬杂质i纯度为99.76%。表3流动相比例time(min)a(%)b(%)0.00208018.00208020.00901040.00901042.002080表4hplc谱图中各峰分析结果peak#rettime(min)areaheightarea%121.6538.250822.293330.2440222.6373373.525391059.2231499.7560totals3381.776211061.51648100.0000实施例2:一种布洛芬杂质i的环保制备方法,包括以下步骤:1)布洛芬杂质h的制备,具体步骤为:步骤1:将0.5g式(i)所示化合物与0.543g式(ii)所示化合物混合溶于etoh中,然后在60-80℃下将0.315gnaoc2h5分4批添加至其中,共搅拌反应15h,将反应液旋蒸发除去etoh,再将得到的固体溶解,经柱层析分离纯化,洗脱剂为pe:ea=8:1,得到式(iii)所示化合物;步骤2:将0.5g式(iii)所示化合物溶于无水thf中,在-20℃下,向其中缓慢加入1.56ml浓度为1mol/l的ch3mgbr试剂,式(iii)所示化合物和ch3mgbr的摩尔比为1:1,在该温度下搅拌反应60min,将反应液旋蒸发除去thf,并通过柱层析分离纯化,洗脱剂为pe:ea=40:1,得到194mg布洛芬杂质h,布洛芬杂质h的产率为37.02%,纯度为99.27%。2)布洛芬杂质i的制备,向含0.3g布洛芬杂质h和0.116gzn的etoh溶液中加入0.130gnh4cl和0.45mlhcl,在70℃搅拌反应12h,反应结果后将反应液旋蒸发除去etoh,再将得到的固体溶解,柱层析分离纯化,洗脱剂为pe:ea=20:1,得到252mg布洛芬杂质i,布洛芬杂质i的产率为85.13%,纯度为99.32%。实施例3:一种布洛芬杂质i的环保制备方法,包括以下步骤:1)布洛芬杂质h的制备,具体步骤为:步骤1:将0.5g式(i)所示化合物与0.543g式(ii)所示化合物混合溶于etoh中,然后在60-80℃下将0.42gnaoc2h5分4批添加至其中,共搅拌反应12h,将反应液旋蒸发除去etoh,再将得到的固体溶解,经柱层析分离纯化,洗脱剂为pe:ea=10:1,得到式(iii)所示化合物;步骤2:将0.5g式(iii)所示化合物溶于无水thf中,在-20℃下,向其中缓慢加入1.56ml浓度为1mol/l的ch3mgbr试剂,以及l-半胱氨酸盐酸盐,式(iii)所示化合物和ch3mgbr的摩尔比为1:1,在该温度下搅拌反应60min,将反应液旋蒸发除去thf,并通过柱层析分离纯化,洗脱剂为pe:ea=50:1,得到230mg布洛芬杂质h,布洛芬杂质h的产率为53.78%,纯度为99.73%;与实施例1相比,本实施例目标产物-布洛芬杂质h的产率得到提高,这可能是因为l-半胱氨酸盐酸盐的引入能够提高格氏试剂ch3mgbr与式(iii)所示化合物(不饱和羰基化合物)反应的产率和选择性,有效抑制副反应的发生,减少醇产物的生产,从而使得目标产物-布洛芬杂质h的产率得到提高,且l-半胱氨酸盐酸盐具有价格低廉和降低污染的优势;2)布洛芬杂质i的制备,向含0.3g布洛芬杂质h和0.116gzn的etoh溶液中加入0.143gnh4cl和0.5mlhcl,在70℃搅拌反应12h,反应结果后将反应液旋蒸发除去etoh,再将得到的固体溶解,柱层析分离纯化,洗脱剂为pe:ea=20:1,得到255mg布洛芬杂质i,布洛芬杂质i的产率为86.34%,纯度为99.81%。实施例4:一种布洛芬杂质i的环保制备方法,包括以下步骤:1)布洛芬杂质h的制备,具体步骤为:步骤1:将0.5g式(i)所示化合物与0.543g式(ii)所示化合物混合溶于etoh中,然后在60-80℃下将0.42gnaoc2h5分4批添加至其中,共搅拌反应12h,将反应液旋蒸发除去etoh,再将得到的固体溶解,经柱层析分离纯化,洗脱剂为pe:ea=10:1,得到式(iii)所示化合物;步骤2:将0.5g式(iii)所示化合物溶于无水thf中,在-20℃下,向其中缓慢加入1.56ml浓度为1mol/l的ch3mgbr试剂,式(iii)所示化合物和ch3mgbr的摩尔比为1:1,在该温度下搅拌反应60min,将反应液旋蒸发除去thf,并通过柱层析分离纯化,洗脱剂为pe:ea=50:1,得到200mg布洛芬杂质h,布洛芬杂质h的产率为38.10%;2)布洛芬杂质i的制备,向含0.3g布洛芬杂质h和0.116gzn的etoh溶液中加入0.143gnh4cl、1.53mg三乙胺和0.5mlhcl,在70℃搅拌反应12h,反应结果后将反应液旋蒸发除去etoh,再将得到的固体溶解,柱层析分离纯化,洗脱剂为pe:ea=20:1,得到266mg布洛芬杂质i,布洛芬杂质i的产率为90.11%,纯度为99.86%。与实施例1相比,本实施例目标产物-布洛芬杂质i的产率得到提高,这可能是因为三乙胺的加入提高了布洛芬杂质h加氢为布洛芬杂质i的活性和选择性,降低了芳环被催化加氢的转化率,提高了布洛芬杂质h中c=o双键的加氢转化率,使得大量的布洛芬杂质h加氢转化为布洛芬杂质i,从而提高目标产物-布洛芬杂质i的产率。实施例5:一种布洛芬杂质i的检测方法,具体包括如下步骤:(1)供试品溶液的配制:精密称取20mg布洛芬原料,加10ml50%乙腈溶液溶解,作为供试品溶液;(2)对照品溶液的配制:精密称取20μg实施例1得布洛芬杂质i,加10ml50%乙腈溶液溶解,作为对照品溶液;(3)样品检测:精密量取上述供试品溶液和对照品溶液各50μl,注入液相色谱仪,色谱条件如下:色谱柱:agilentec-c184.6×150mm2.7μm;流速:0.3ml/min;检测波长:254nm;进样量:20μl;柱温:25℃;流动相:a:95%乙腈;b:0.1%甲酸溶液;按表5进行梯度洗脱程序,记录色谱图。表5流动相比例time(min)a(%)b(%)0.00208018.00208020.00901040.00901042.002080实施例6:一种布洛芬杂质i的检测方法,与实施例4的不同之处在于,对照品溶液的配制用实施例2得布洛芬杂质i。实施例7:一种布洛芬杂质i的检测方法,与实施例4的不同之处在于,对照品溶液的配制用实施例3得布洛芬杂质i。实施例8:一种布洛芬杂质i的检测方法,与实施例4的不同之处在于,对照品溶液的配制用实施例4得布洛芬杂质i。试验例1:检测方法的色谱系统方法学验证1.检测限和定量限取布洛芬供试品溶液及布洛芬杂质i对照品溶液,分别稀释成一系列溶度,在上述实施例4“(3)样品检测”色谱条件下进样,记录色谱图,以主成分峰高约为基线噪音的3倍计算各杂质的检测限,以主成分峰高约为基线噪音的10倍计算各杂质的定量限,见表6。2.线性关系取布洛芬杂质i对照品溶液,配制成不同质量浓度的一系列线性溶液,在上述实施例4“(3)样品检测”色谱条件下分别进样,记录色谱图。以进样质量浓度(g/l)为横坐标,峰面积为纵坐标,进行线性回归分析,实施例5、实施例6、实施例7和实施例8布洛芬中布洛芬杂质i在一定质量浓度范围内线性相关性良好,其线性范围见表6。表6检测方法的检测限、定量限、线性范围、回归方程和相关系数组别检测限(mg/l)定量限(mg/l)线性范围(mg/l)回归方程r2实施例564.33112.670.446-1.705y=2.85×104x-3.14×10364.33实施例666.07114.940.472-1.689y=2.83×104x-3.16×10366.07实施例763.03110.040.429-1.713y=2.87×104x-3.10×10363.03实施例862.96110.010.430-1.715y=2.87×104x-3.09×10362.963.重复性取系统适用性溶液,在实施例4“(3)样品检测”色谱条件下分别连续进样6次,记录峰面积计算rsd值,实施例5、实施例6、实施例7和实施例8布洛芬杂质i的rsd%分别为0.32%、0.35%、0.31%和0.31%(n=6)。结果表明:实施例5、实施例6、实施例7和实施例8方法精密度良好,(rsd%≤5.0%)。4.溶液稳定性试验取对照品溶液和供试品溶液,室温放置,分别于0、2、4、6、8、10、12h、16h取上述对照品溶液和供试品溶液,在实施例4“(3)样品检测”色谱条件下分别进样,实施例5、实施例6、实施例7和实施例8布洛芬杂质i峰面积的rsd%分别为:0.52%,0.55%,0.48%,0.49%。结果表明:对照品溶液和供试品溶液在配制16h内稳定。5.准确度试验采用加样回收试验,配制加样浓度分别为限度浓度80%、100%和120%水平的供试溶液,每个水平平均配制9份。在实施例4“(3)样品检测”色谱条件下分别进样,记录色谱图。实施例5、实施例6、实施例7和实施例8测得布洛芬杂质i的平均回收率分别为99.97%、99.91%、99.98%。结果表明:布洛芬杂质i的平均回收率至少为99.90%,rsd%≤5.0%,说明本方法的准确度良好。上述实施例中的常规技术为本领域技术人员所知晓的现有技术,故在此不再详细赘述。以上实施方式仅用于说明本发明,而并非对本发明的限制,本领域的普通技术人员,在不脱离本发明的精神和范围的情况下,还可以做出各种变化和变型。因此,所有等同的技术方案也属于本发明的范畴,本发明的专利保护范围应由权利要求限定。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1