一种含C(sp2)-S键芳基砜化合物的合成方法与流程
本发明涉及有机合成领域,具体涉及一种含c(sp2)-s键芳基化合物的合成方法。
背景技术:
砜类基团是有机分子中常见的结构单元之一,广泛存在于药物分子和功能性材料中,其在合成化学中也起着重要作用,例如经典的兰堡–巴克伦(ramberg-backlund)反应和朱利亚烯烃合成(juliaolefination)反应。近年来对砜类化合物的合成研究也成为一个热门,传统的合成方法主要包括硫化物或亚砜类的氧化,在强酸条件下芳烃的磺酰化,或是将磺酰基自由基前体加成到烯烃和炔烃中去。然而,这些方法通常存在需要多步预官能化反应、区域选择性差或官能团相容性差的问题。如下述非专利文献报道:a)s.patai,z.rappoport,c.j.m.stirling,thechemisttryofsulfonesandsulfoxides,wiley,newyork,1988;b)q.a.acton,sulfones–advancesinresearchandapplication,scholarlyeditions,atlanta,2013;c)l.ramberg,b.b-cklund,ark.chim.mineralgeol.1940,27,1–50;d)s.c.
近年来,有机合成研究中可以通过各种过渡金属(例如pd,cu,rh和ru)作为催化剂,在其催化下通过c–h键活化的偶联反应合成各类化合物,过渡金属催化的c-h键介导已成为合成碳-碳和碳-杂原子键的有力工具,在这种策略的启发下,我们可以通过c–h键的直接磺酰化合成砜类化合物。如下述非专利文献报道:a)r.giri,b.-f.shi,k.m.engle,n.maugelandj.-q.yu,chem.soc.rev.,2009,38,3242;(b)l.mcmurray,f.o.haraandm.j.gaunt,chem.soc.rev.,2011,40,1885;(c)j.wenceldelordandf.glorius,nat.chem.,2013,5,369;e)x.chen,k.m.engle,d.-h.wangandj.-q.yu,angew.chem.,int.ed.,2009,48,5094;f)d.a.colby,r.g.bergmanandj.a.ellman,chem.rev.,2010,110,624;g)p.b.arockiam,c.bruneauandp.h.dixneuf,chem.rev.,2012,112,5879;h)s.shaaban,s.liang,n.-w.liuandg.manolikakes,org.biomol.chem.,2017,15,1947;i)n.-w.liu,s.liangandg.manolikakes,synthesis,2016,1939.;j)xia,h.;an,y.;zeng,x.;wu,j.,palladium-catalyzeddirectsulfonylationofc-hbondswiththeinsertionofsulfurdioxide.chemcommun(camb)2017,53(93),12548-12551。沿着这一思路,dong在2009年开发了一种钯催化2-芳基吡啶与芳基磺酰氯的直接c–h键磺酰化反应,但其底物类型相当有限(x.zhao,e.
技术实现要素:
本发明旨在至少解决现有技术中存在的技术问题之一。为此,本发明提出一种含c(sp2)-s键芳基砜化合物的合成方法,能够通过空气氧化实现c(sp2)-h键的磺酰化反应。
根据本发明的第一方面实施例的一种含c(sp2)-s键芳基砜化合物的合成方法,包括以下步骤:以如式(i)所示的5-氯-8-氨基喹啉芳族酰胺类化合物与如式(ii)所示的芳基亚磺酸盐在铜催化剂、碱试剂和配位苯甲酸存在的条件下反应,即得到如式(ⅲ)所示的芳基砜化合物:
式中,环a代表未取代或取代的c6~c10芳基、未取代或取代的“杂原子”为s的或杂原子数为1~4个的c3~c8杂芳基;所述环a为式(i)中所示的
根据本发明实施例的合成方法,至少具有如下有益效果:本发明的方法是以底物的5-氯-8-氨基喹啉部分作为导向基团通过空气氧化实现c(sp2)-h键的磺酰化反应;该方法原料简单易得,反应操作简便,不需要使用其他的氧化剂,环境友好,利于工业化应用;该方法的选择性高、官能团相容性好,为合成芳基磺酰类化合物提供了一种简洁高效的制备方法。
作为优选的,所述c6~c10芳基为苯基或萘基
所述含1~4个s或o原子的c3~c8杂芳基为噻吩基
作为优选的,所述环a为取代苯基
作为优选的,r1可为甲基、苯基、甲氧基、卤素(如氯、溴等)、三氟甲基、羟基等;r2可为甲基、卤素(如氟)、三氟甲基、氰基等;r3可为甲基、乙基、叔丁基、苯基、卤素(如氟、氯)、甲磺酰基等;r1和r3,r2和r3均可同时为甲基等。
根据本发明的一些实施例,所述环a为如下任一结构:
作为优选的,所述环a为如下任一结构:
作为优选的,所述环b为取代苯基
作为优选的,r2可为卤素(如氯)等;r3可为甲基、叔丁基、卤素(如氟、氯)、甲氧基、卤代烷基(如三氟甲基)等;r1和r3均可同时为卤素等。
根据本发明的一些实施例,所述环b为如下任一结构:
根据本发明的一些实施例,所述5-氯-8-氨基喹啉芳族酰胺类化合物为如下任一结构:
根据本发明的一些实施例,所述芳基亚磺酸盐类化合物为如下任一结构:
根据本发明的一些实施例,所述芳基砜化合物为如下任一结构:
根据本发明的一些实施例,所述cu催化剂包括一价铜盐和二价铜盐中的至少一种;
作为优选的,所述一价铜盐包括cucl、cubr、cui和cu2o中的至少一种;
作为优选的,所述二价铜盐包括cu(oac)2·h2o、cu(oac)2、cu(no3)2·3h2o、cucl2、cubr2和cu(otf)2中的至少一种。
作为优选的,所述cu催化剂为cu(oac)2。
根据本发明的一些实施例,所述碱试剂包括碱金属的有机酸盐和碱金属的无机酸盐中的至少一种;作为优选的,所述碱试剂包括kopiv、koac、naoac、naopiv·h2o和k2co3中的至少一种。
作为优选的,所述碱试剂为kopiv或naoac中的至少一种。
根据本发明的一些实施例,反应使用的溶剂包括dmf、dma、thf、dmso、dce、乙腈和1,4-二氧六环中的至少一种。
作为优选的,所述溶剂包括dmf和dma中的至少一种。
根据本发明的一些实施例,所述配位苯甲酸包括苯甲酸、间甲基苯甲酸、3,4-二甲基苯甲酸、2,4,6-三甲基苯甲酸、4-叔丁基苯甲酸、邻甲基苯甲酸和2-苯基苯甲酸中的至少一种。
作为优选的,所述配位苯甲酸包括苯甲酸和间甲基苯甲酸中的至少一种。
根据本发明的一些实施例,所述5-氯-8-氨基喹啉芳族酰胺类化合物、芳基亚磺酸钠、cu催化剂、配位苯甲酸和碱试剂的摩尔比为1:(1~4):(0.05~0.4):(0.1~5):(1~4)。
作为优选的,所述5-氯-8-氨基喹啉芳族酰胺类化合物、芳基亚磺酸钠、cu催化剂、配位苯甲酸和碱试剂的摩尔比为1:2:0.2:0.3:2。
根据本发明的一些实施例,反应所选温度为(70~120)℃;反应时间为(4~12)h。
作为优选的,所述温度为80℃;所述反应时间为6h。
本发明中,术语“取代”的位置,如未做特别说明,位置可为任意;例如各自独立地位于“芳基与其它基团连接位点”、或“杂芳基与其它基团连接位点”的邻位、间位或对位,以苯基为例,是指取代基位于中键的邻位、间位或对位。
本发明中,如未做特别说明,卤素包括f、cl、br或i。
本领域技术人员可以理解,根据本领域中使用的惯例,本申请描述基团的结构式中所使用的
本发明的附加方面和优点将在下面的描述中部分给出,部分将从下面的描述中变得明显,或通过本发明的实践了解到。
附图说明
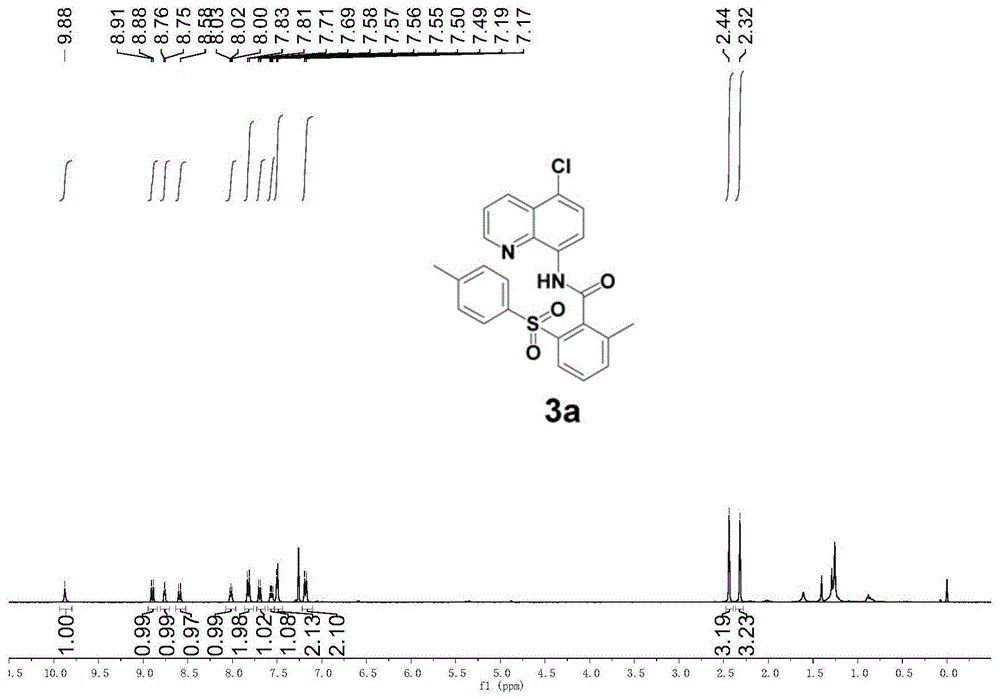
图1为本发明实施例1制得的化合物的1hnmr谱图;
图2为本发明实施例1制得的化合物的13cnmr谱图;
图3为本发明实施例2制得的化合物的1hnmr谱图;
图4为本发明实施例2制得的化合物的13cnmr谱图;
图5为本发明实施例3制得的化合物的1hnmr谱图;
图6为本发明实施例3制得的化合物的13cnmr谱图;
图7为本发明实施例4制得的化合物的1hnmr谱图;
图8为本发明实施例4制得的化合物的13cnmr谱图;
图9为本发明实施例5制得的化合物的1hnmr谱图;
图10为本发明实施例5制得的化合物的13cnmr谱图;
图11为本发明实施例6制得的化合物的1hnmr谱图;
图12为本发明实施例6制得的化合物的13cnmr谱图。
具体实施方式
为详细说明本发明的技术内容、所实现目的及效果,以下结合实施方式并配合附图予以说明。
实施例1
在10ml反应管中依次加入搅拌子、dmf(1ml)、1a(59.4mg,0.2mmol)、2a(106.9mg,0.6mmol)、醋酸铜(ii)(7.2mg,0.02mmol)、苯甲酸(7.4mg,0.03mmol)、特戊酸钾(56mg,0.4mmol),反应体系置于80℃油浴中反应8h。反应结束后冷却至室温,用乙酸乙酯(5ml)稀释后,用水(3×20ml)洗涤,再用盐水(20ml)洗涤,合并有机相并用无水na2so4干燥,过滤得滤液浓缩,硅胶柱层析(石油醚:乙酸乙酯)分离得到白色固体3a(72.2mg,80%,纯度大于95%)。该反应产率高达80%,使用简单的分离纯化方法即可得到纯度大于95%的产物,说明该反应方法具有反应产率高和选择性好等优点。
产品检测数据如下:1hnmr(400mhz,cdcl3)δ9.88(s,1h),8.90(d,j=8.4hz,1h),8.76(d,j=3.9hz,1h),8.59(d,j=8.4hz,1h),8.07–7.96(m,1h),7.82(d,j=8.1hz,2h),7.70(d,j=8.3hz,1h),7.56(dd,j=8.5,4.1hz,1h),7.50(d,j=4.9hz,2h),7.18(d,j=8.0hz,2h),2.44(s,3h),2.32(s,3h)。
13cnmr(101mhz,cdcl3)δ165.46,148.75,144.18,139.00,138.77,138.23,137.16,136.06,135.45,133.55,133.32,129.62,128.13,127.22,127.04,125.98,124.97,123.99,122.37,116.87,21.56,19.27。
实施例2
在10ml反应管中依次加入搅拌子、dmf(1ml)、1a(72.3mg,0.2mmol)、2a(106.9mg,0.6mmol)、醋酸铜(ii)(7.2mg,0.02mmol)、苯甲酸(7.4mg,0.03mmol)、特戊酸钾(56mg,0.4mmol),反应体系置于80℃油浴中反应8h。反应结束后冷却至室温,用乙酸乙酯(5ml)稀释后,用水(3×20ml)洗涤,再用盐水(20ml)洗涤,合并有机相并用无水na2so4干燥,过滤得滤液浓缩,硅胶柱层析(石油醚:乙酸乙酯)分离得到白色固体3b(64.5mg,63%,纯度大于95%)。
产品检测数据如下:1hnmr(400mhz,cdcl3)δ10.03(s,1h),8.82(d,j=8.4hz,1h),8.78(d,j=4.0hz,1h),8.58(d,j=8.5hz,1h),8.21(d,j=7.5hz,1h),7.86(d,j=8.1hz,2h),7.68–7.63(m,3h),7.56(dd,j=8.5,4.2hz,1h),7.21(d,j=8.2hz,2h),2.32(s,3h)。13cnmr(101mhz,cdcl3)δ165.68,148.81,144.34,139.00,138.94,137.91,136.80,133.64,133.45,133.28,130.39,129.70,129.61,128.66,128.32,127.13,125.91,125.03,122.44,116.87,21.57。
实施例3
在10ml反应管中依次加入搅拌子、dmf(1ml)、1a(71.8mg,0.2mmol)、2a(106.9mg,0.6mmol)、醋酸铜(ii)(7.2mg,0.02mmol)、苯甲酸(7.4mg,0.03mmol)、特戊酸钾(56mg,0.4mmol),反应体系置于80℃油浴中反应8h。反应结束后冷却至室温,用乙酸乙酯(5ml)稀释后,用水(3×20ml)洗涤,再用盐水(20ml)洗涤,合并有机相并用无水na2so4干燥,过滤得滤液浓缩,硅胶柱层析(石油醚:乙酸乙酯)分离得到白色固体3c(52.41mg,51%,纯度大于95%)。
产品检测数据如下:1hnmr(400mhz,cdcl3)δ9.59(s,1h),8.60(d,j=8.1hz,2h),8.44(d,j=8.5hz,1h),8.23(d,j=6.7hz,1h),7.92(d,j=8.1hz,2h),7.63(s,2h),7.54(d,j=8.3hz,1h),7.41(d,j=8.1hz,3h),7.25–7.19(m,2h),7.16(t,j=7.4hz,2h),7.09(d,j=7.3hz,1h),2.29(s,3h)。
13cnmr(101mhz,cdcl3)δ164.67,148.45,144.25,141.61,139.59,138.74,138.32,138.21,135.36,135.24,133.44,133.01,129.73,129.58,128.73,128.47,128.16,128.01,127.04,125.67,124.56,122.16,116.44,21.58。
实施例4
在10ml反应管中依次加入搅拌子、dmf(1ml)、1a(62.2mg,0.2mmol)、2a(106.9mg,0.6mmol)、醋酸铜(ii)(7.2mg,0.02mmol)、苯甲酸(7.4mg,0.03mmol)、特戊酸钾(56mg,0.4mmol),反应体系置于80℃油浴中反应8h。反应结束后冷却至室温,用乙酸乙酯(5ml)稀释后,用水(3×20ml)洗涤,再用盐水(20ml)洗涤,合并有机相并用无水na2so4干燥,过滤得滤液浓缩,硅胶柱层析(石油醚:乙酸乙酯)分离得到白色固体3d(72.63mg,78%,纯度大于95%)。
产品检测数据如下:1hnmr(400mhz,cdcl3)δ9.84(s,1h),8.89(d,j=8.4hz,1h),8.72(d,j=2.8hz,1h),8.55(d,j=8.5hz,1h),7.83(d,j=8.5hz,3h),7.68(d,j=8.4hz,1h),7.53(dd,j=8.5,4.2hz,1h),7.28(s,1h),7.17(d,j=8.1hz,2h),2.41(s,3h),2.39(s,3h),2.30(s,3h)。
13cnmr(101mhz,cdcl3)δ165.75,148.72,144.07,140.02,138.94,138.54,138.36,136.98,136.14,133.64,133.51,133.24,129.60,128.07,127.28,127.20,125.92,124.83,122.36,116.75,21.55,21.19,19.19。
实施例5
在10ml反应管中依次加入搅拌子、dmf(1ml)、1a(59.4mg,0.2mmol)、2b(198.60mg,0.6mmol)、醋酸铜(ii)(7.2mg,0.02mmol)、苯甲酸(7.4mg,0.03mmol)、特戊酸钾(56mg,0.4mmol),反应体系置于80℃油浴中反应8h。反应结束后冷却至室温,用乙酸乙酯(5ml)稀释后,用水(3×20ml)洗涤,再用盐水(20ml)洗涤,合并有机相并用无水na2so4干燥,过滤得滤液浓缩,硅胶柱层析(石油醚:乙酸乙酯)分离得到白色固体3a(60.33mg,64%,纯度大于95%)。
产品检测数据如下:1hnmr(400mhz,chloroform-d)δ9.90(s,1h),8.89(d,j=8.3hz,1h),8.76(dd,j=4.3,1.6hz,1h),8.58(dd,j=8.5,1.6hz,1h),8.02(t,j=4.6hz,1h),8.00–7.91(m,2h),7.69(d,j=8.4hz,1h),7.56(dd,j=8.5,4.2hz,1h),7.51(d,j=4.7hz,2h),7.40(t,j=7.7hz,2h),2.45(s,3h)。
13cnmr(101mhz,cdcl3)δ165.60,148.94,141.41,139.16,138.63,137.38,136.38,135.79,133.68,133.50,133.36,129.82,129.14,128.23,127.37,126.15,125.18,122.56,117.07,19.44。
实施例6
在10ml反应管中依次加入搅拌子、dmf(1ml)、1a(59.4mg,0.2mmol)、2c(220.26mg,0.6mmol)、醋酸铜(ii)(7.2mg,0.02mmol)、苯甲酸(7.4mg,0.03mmol)、特戊酸钾(56mg,0.4mmol),反应体系置于80℃油浴中反应8h。反应结束后冷却至室温,用乙酸乙酯(5ml)稀释后,用水(3×20ml)洗涤,再用盐水(20ml)洗涤,合并有机相并用无水na2so4干燥,过滤得滤液浓缩,硅胶柱层析(石油醚:乙酸乙酯)分离得到白色固体3a(60.15mg,80%,纯度大于95%)。
产品检测数据如下:1hnmr(400mhz,chloroform-d)δ9.91(s,1h),8.92(d,j=8.4hz,1h),8.78–8.68(m,1h),8.60–8.47(m,1h),8.00(dd,j=6.6,2.7hz,1h),7.91(d,j=8.2hz,2h),7.69(d,j=8.4hz,1h),7.53(dd,j=8.5,4.2hz,1h),7.49–7.44(m,2h),7.44–7.37(m,2h),2.43(s,3h),1.25(s,9h)。
13cnmr(101mhz,cdcl3)δ165.70,157.11,148.87,139.03,138.92,138.28,137.08,136.10,135.52,133.64,133.33,129.75,128.07,127.22,126.15,126.01,125.02,122.50,116.96,35.17,31.03,19.35。
综上所述,本发明提供的芳基砜化合物的合成方法,以底物的5-氯-8-氨基喹啉部分作为导向基团通过空气氧化实现c(sp2)-h键的磺酰化反应,反应选择性高且官能团相容性好;该方法原料简单易得,反应操作简便,不需要使用其他的氧化剂,环境友好,利于工业化应用,为合成芳基磺酰类化合物提供了一种简洁高效的制备方法。
以上所述仅为本发明的实施例,并非因此限制本发明的专利范围,凡是利用本发明说明书及附图内容所作的等同变换,或直接或间接运用在相关的技术领域,均同理包括在本发明的专利保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!