一种L-赖氨酸选择性结构修饰的方法与流程
本发明属于化学合成领域,具体涉及一种l-赖氨酸选择性结构修饰的方法。
背景技术:
氨基酸的选择性保护是合成定向肽的重要手段。对于包含多功能组的氨基酸,如赖氨酸、酪氨酸,精氨酸等,应采用不同的保护方法、以有选择地保护这些氨基酸的活性官能团,从而达到氨基酸的方向选择性偶联反应。其中,l-赖氨酸作为一种必需氨基酸,在多肽合成中起着重要作用。然而,由于赖氨酸的两个氨基具有基本相同的化学性质,很难控制反应的选择性,从而难以得到保护侧链氨基的单一赖氨酸。
目前,文献报道的l-赖氨酸选择性保护的方法主要有:(1)在酸性条件下,通过硫酸铜与l-赖氨酸的α-nh2络合,暂时保护α-nh2,再将赖氨酸络合铜加入水与四氢呋喃的混合溶剂中,加入氢氧化钠或碳酸钠将溶液调整至碱性,在冰浴条件下逐滴加入氯甲酸苄酯进行反应。反应结束后添加脱铜试剂以脱除络合态的铜离子,得到终产物。该方法有较高产率,生产成本低廉,但在生产过程中会产生大量含有重金属铜的废水,造成环境污染问题。(2)以β-环糊精为催化剂,以水作溶剂,将l-赖氨酸与氯甲酸苄酯在室温下反应,得到终产物(例如:org.biomol.chem.,2012,10,9319–9324。中国专利:一种赖氨酸氨基的辛氧裁基高效选择性保护方法及其产品,申请号201110422510.4)。环糊精具有外亲水内疏水的特性,能够为反应提供独特的微环境,因此该方法反应速率快,产物产率高,反应条件温和。但由于反应在环糊精疏水空腔内进行,反应后产物具有较强的疏水性,导致产物与β-环糊精难以在水溶液中直接分离。按照文中描述,该方法的后续分离处理较为容易,只需加入乙酸乙酯萃取处理后即得产物。然而发明人在实际操作中发现:在反应结束后,环糊精与产物的混合物浮于反应液表面,呈白色固体状。按照文献中的萃取方法处理后,所得仍为环糊精与产物的混合物,无法实现分离产物与环糊精的目的。发明人进一步优化该方法的后处理方式:加入乙酸乙酯至反应液中,超声处理五分钟,大力搅拌一小时,静置分层后,分离出产物。但这种优化的后处理方法需要使用大量挥发性有机溶剂,后处理操作复杂、纯化难度大。
因此,提供一种后处理操作简便、分离效果优异、有机溶剂用量少的l-赖氨酸选择性结构修饰方法,具有重要的科研价值和现实意义。
技术实现要素:
本发明的目的是提供一种l-赖氨酸选择性结构修饰的方法。
为实现上述发明目的,本发明所采用的技术方案是:一种用于l-赖氨酸选择性保护反应的催化剂,所述催化剂为β-环糊精表面功能化的二氧化硅纳米粒子。
优选的,所述催化剂粒径为100nm。
优选的,所述催化剂中,β-环糊精的质量百分比为10%±0.5%。
相应的,所述催化剂的制备方法包括如下步骤:
(1)通过β-环糊精的6位羟基与异氰酸丙基三乙氧基硅烷的异氰酸基团进行偶联反应,获得功能单体cdsi;
(3)将球状二氧化硅纳米粒子与所述cdsi以质量比为1:1的比例添加到无水dmf中,在惰性气氛下110℃反应、过滤、洗涤、干燥,得所述催化剂。
优选的,所述球状二氧化硅纳米粒子的粒径为20~200nm。
相应的,一种选择性修饰l-赖氨酸的方法,以l-赖氨酸为原料,以所述β-环糊精表面功能化的二氧化硅纳米粒子为催化剂,在氨基保护剂的存在下进行反应。
优选的,所述氨基保护剂为氯甲酸苄酯、苄溴、苄氯、9-芴甲基-n-琥珀酰亚胺基碳酸酯、芴甲氧羰酰氯、烯丙基氯甲酸酯、二碳酸二叔丁酯、对甲基苯磺酰氯、对硝基苯磺酰氯、对甲氧基氯化苄、三苯甲基溴中的任意一种。
优选的,所述方法包括如下步骤:
(1)以l-赖氨酸为原料,以β-环糊精表面功能化的二氧化硅纳米粒子为催化剂,以水为溶剂,在氨基保护剂的存在下,在25℃下反应3~5小时,得反应终产物;
(2)将所述反应终产物静置,取上层产物,洗涤、分离、过滤,得选择性修饰的l-赖氨酸。
优选的,所述l-赖氨酸与β-环糊精表面功能化的二氧化硅纳米粒子的质量比为1:3.2。
优选的,所述l-赖氨酸与氨基保护剂的摩尔比为1:1。
本发明具有以下有益效果:本发明首次使用β-环糊精表面功能化的二氧化硅纳米材料为催化剂进行l-赖氨酸的选择性结构修饰反应。所述催化剂的原料价廉易得,合成方法简单;合成的催化剂无味、无毒,易于产物分离、可重复利用。该催化剂不仅保留了β-环糊精良好催化性能,同时引入了具有亲水性的纳米粒子,反应结束后,经修饰的l-赖氨酸具有疏水性,催化剂的纳米粒子表面具有亲水性,两者极易分离。从而有效解决了普通β-环糊精催化后产物与环糊精混合物浮于液面而难分离的问题,简化了后处理步骤,减少了有机溶剂的用量,大幅减少了纯化阶段产生的废液量。
附图说明
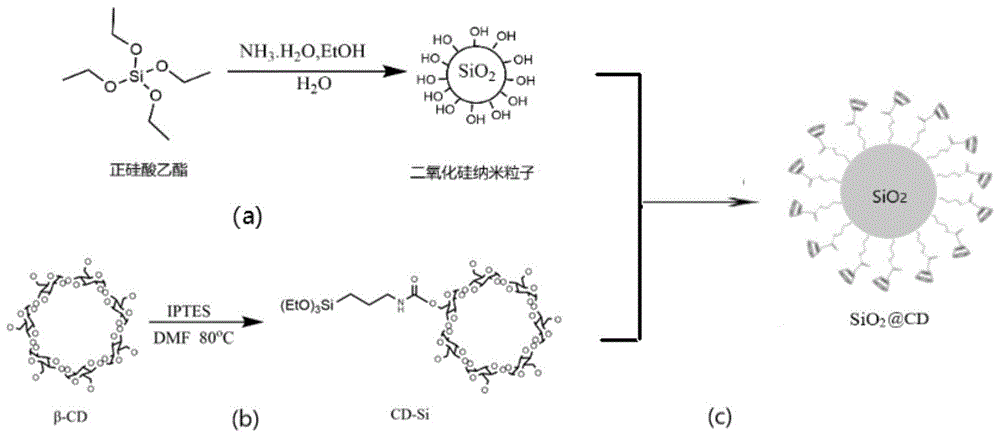
图1为β-环糊精表面功能化二氧化硅纳米粒子的制备方法示意图;
图2为cdsi的核磁氢谱鉴定示意图;
图3为sio2纳米粒子和sio2@cd的tga曲线;
图4为sio2纳米颗粒的电镜扫描图。
具体实施方式
一、本发明提供了一种用于l-赖氨酸选择性保护反应的催化剂。具体为一种β-环糊精表面功能化的二氧化硅纳米粒子(全文简称为:sio2@cd)。所述sio2@cd的粒径为20~200nm,优选为100nm。所述sio2@cd中,β-环糊精的质量百分比为10%±0.5%。
所述sio2@cd的制备过程如图1所示,具体包括如下步骤:
(1)采用
(2)通过β-环糊精(β-cd)的6位羟基与异氰酸丙基三乙氧基硅烷(iptes)的异氰酸基团进行偶联反应。具体方法为:将β-环糊精与iptes以摩尔比1:1.5的比例溶于无水n,n-二甲基甲酰胺(dmf)中,在惰性气氛下80℃反应6小时,反应结束后,冷却至室温,加入丙酮沉淀纯化,过滤,得白色固体产物,即功能单体cdsi。
为确定β-环糊精与iptes的偶联反应是否成功,通过400m核磁氢谱进行鉴定分析,图谱如图2所示。结果为:1hnmr(dmso)dppm0.48-0.87(4h,m,ch2ch2si);1.15-1.60(9h,m,(ch3)3);3.15-3.18(2h,m,ch2);3.56-3.62(6h,m,(ch2)3);4.13-4.16(2h,m,ch2);4.48(h,m,oh);4.83(h,d,oh);5.68-5.74(2h,m,oh);7.95(h,s,nh)。将该谱图与未修饰的环糊精进行对比发现:在δ0.48-0.87区域出现硅烷基团(ch2ch2si)的峰;在δ4.48处,6位羟基氢峰值比例降低;在δ4.13-4.16处出现新的峰,该峰归属为6位羟基被iptes取代后的6位烷基氢的峰。结合各峰积分面积的比例,确定该化合物为β-环糊精与功能单体的复合物,即cdsi。
(3)将步骤(1)的sio2nps与步骤(2)的cdsi以质量比为1:1的比例添加到无水dmf中,在惰性气氛下110℃反应24小时。过滤洗涤干燥后,即得到β-环糊精表面功能化二氧化硅纳米粒子sio2@cd,所得sio2@cd中β-环糊精质量百分比为10%±0.5%。
具体可采用热失重法测定sio2@cd中β-环糊精质量百分比含量。本示例中,sio2纳米粒子和sio2@cd的tga曲线如图3所示。sio2@cd的tga曲线包括两次降解。第一次降解范围为200℃到450℃,为有机物的降解阶段。通过与sio2纳米粒子的热失重结果进行对比发现:sio2@cd表面有机物含量为11%,从而计算出其表面有机物中β-环糊精占比为0.93,由此得出:sio2@cd表面的β-环糊精含量为10.2%。
二、本发明基于所述sio2@cd,还提供了一种选择性修饰l-赖氨酸的方法。反应方程式为:
具体为:
(1)以l-赖氨酸为原料,以所述sio2@cd为催化剂,以a为氨基保护剂,以水为溶剂,在温度为25℃的反应条件下,反应3~5小时,即得反应终产品。
(2)将所述反应终产物移至分液漏斗,静置1h后,产物浮于上层。取上层产物,加水洗涤分离3次后,过滤,再用乙酸乙酯洗涤过滤,即得产物b。当生成的终产物与副产物不易直接分离时,进一步将产物b再通过高效液相色谱法分离,得提纯、去除副产物后的产物b。
其中,氨基保护剂a为氯甲酸苄酯、苄溴、苄氯、9-芴甲基-n-琥珀酰亚胺基碳酸酯、芴甲氧羰酰氯、烯丙基氯甲酸酯、二碳酸二叔丁酯、对甲基苯磺酰氯、对硝基苯磺酰氯、对甲氧基氯化苄、三苯甲基溴中任意一种。产物b中r基团为苄氧羰基、苄基、芴甲氧羰基、烯丙氧羰基、叔丁氧羰基、对甲苯磺酰基、硝基苯磺酰基、对甲氧基苄基、三苯甲基中的任意一种,r基团具体为与a试剂相对应的基团。
优选的方案为:步骤(1)中,l-赖氨酸与sio2@cd的质量比为1:3.2;l-赖氨酸与a的摩尔比为1:1。
下面结合具体的实施例,对本发明进行进一步阐释。
实施例一:制备sio2@cd
1、制备大小均匀的sio2纳米粒子。将乙酸乙酯(44ml)、h2o(11ml)和氨水(1.1ml)加入锥形瓶中,在40℃下搅拌混合,转速调节至300rpm/min。随后将teos(4.4ml)和无水乙醇(20ml)均匀混合,加入上述反应体系中,加入完毕后,转速调至600rpm/min,搅拌3min。而后将转速降至300rpm/min,搅拌15h。所得液体经高速离心(9000rpm/min),多次洗涤后获得sio2纳米颗粒(sio2nps),最后将其在120℃下干燥24h,密封干燥储存待用。sio2nps的电镜扫描图如图4所示。
2、制备sio2@cd。将β-cd在100℃、真空条件下干燥24h,随后取β-cd(0.55mmol)溶于的无水dmf(10ml)中。随后逐滴加入iptes(0.15ml)。滴加完成后,在惰性气体条件下,将温度升至80℃反应6h。反应结束后,冷却至室温,加入丙酮沉淀纯化,过滤,得白色固体产物,即为β-cd功能单体复合物(cdsi)。然后将cdsi(500mg)与sio2nps(500mg)加入至无水dmf(30ml)中,在惰性气体下110℃剧烈搅拌反应24h。过滤,洗涤,材料干燥12h,即得sio2@cd。
实施例二:制备一种选择性结构修饰后的l-赖氨酸
本实施例涉及反应方程式为:
将sio2@cd(40mg)加入到碳酸钠水溶液(4ml)中,ph值约为9.5。搅拌至均匀分散后,加入l-赖氨酸(0.1mmol),继续搅拌1h后,逐滴加入的a1(氯甲酸苄酯,cbz-cl,0.09mmol)。反应3h后,停止反应,将反应液移至分液漏斗,静置1h,取上层产物,加水洗涤分离3次后,过滤,再用乙酸乙酯(3ml)洗涤,得终产物b1,即nε-cbz-l-赖氨酸,产率为78.6%,其选择性>99%。
b1为白色固体。谱图数据为:1hnmr(dmso)dppm1.13-1.45(4h,m,ch2ch2);1.48-1.76(2h,m,ch2);2.90-3.01(2h,m,ch2ε);3.02-3.08(h,t,ch);5.00(2h,s,ch2ar);7.23-7.40(5h,m,ar)。13c-nmr(dmso)δ171.38,156.56,137.69,128.83,128.22,128.18,65.60,52.26,30.02,29.28,22.02。msm/z:281.1563(m+)。
实施例三:制备另一种选择性结构修饰后的l-赖氨酸
本实施例涉及反应方程式为:
将sio2@cd(100mg)加入到碳酸钠水溶液(10ml)中,ph值约为10.5。搅拌至均匀分散后,加入l-赖氨酸(0.1mmol),继续搅拌1h后,逐滴加入的a2(苄溴,brbn,0.09mmol),反应5h后,停止反应。将反应液移至分液漏斗,静置1h后,取上层产物,用水洗涤分离三次后,过滤,再用乙酸乙酯(3ml)洗涤,得粗产物。将所述粗产物通过高效液相分离,得终产物b2,即nε-bn-l-赖氨酸,产率为46.6%,选择性>99%。高效液相色谱分离条件为:流动相:acn/h2o(40/60),紫外波长:208nm,流速:1ml/min,每针进样25μl。
b2为白色固体。谱图数据为:1hnmr(dmso)dppm1.21-1.50(6h,m,ch2ch2ch2);2.97-3.15(2h,m,ch2ε);3.20-3.25(h,t,ch);5.00(2h,s,ch2ar);7.23-7.48(5h,m,ar)。13cnmr(dmso)δ173.45,140.03,128.91,128.62,128.21,57.93,53.13,50.77,31.86,26.65,23.64。msm/z:237.1553(m+)。
实施例四:制备第三种选择性结构修饰后的l-赖氨酸
本实施例涉及反应方程式为:
将sio2@cd(100mg)加入到碳酸钠水溶液(10ml)中,ph值约为9。搅拌至均匀分散后,加入l-赖氨酸(0.1mmol),继续搅拌1h后,逐滴加入a3(9-芴甲基-n-琥珀酰亚胺基碳酸酯,fmoc-osu,0.09mmol),反应5h后,停止反应。因本发明制备获得的产物在ph>3时难溶于乙酸乙酯,在ph为2~3时可溶于乙酸乙酯,故用稀盐酸将ph值调至2~3,随后再将产物溶于乙酸乙酯(3ml),过滤,取滤液。高效液相色谱分离,得终产物b3,即nε-fmoc-l-赖氨酸,产率为18.3%,选择性>97%。高效液相色谱分离条件为:流动相:acn/h2o(35/65),紫外波长:208nm,流速:1ml/min,每针进样25μl。
b3为白色固体。谱图数据为:1hnmr(dmso)dppm1.23-1.38(2h,m,ch2);1.56-1.69(2h,m,ch2);1.69-1.72(2h,m,ch2);2.96-2.97(h,m,ch);3.12(2h,m,ch,ch);4..21-4.23(h,m,ch);4.38-4.30(2h,m,ch2);7.32-7.90(8h,m,ar)。13cnmr(dmso)δ171.26,156.53,144.01,141.30,128.10,127.70,125.62,120.52,65.56,52.32,46.98,29.80,29.08,21.92。msm/z:369.1861(m+)。
- 还没有人留言评论。精彩留言会获得点赞!