以三聚喹唑啉为结点的二维共价有机框架材料及其制备方法和应与流程
本发明属于多孔有机高分子领域,涉及一种二维共价有机框架材料,具体地说是一种以三聚喹唑啉为结点的二维共价有机框架材料及其制备方法和应用。
背景技术:
二维共价有机框架材料(cofs)是一类长程有序的多孔结晶材料,它具有高度有序、孔径可调、比表面积较大、合成方法多样和易于功能化修饰等优点,是一类新兴的多相催化剂。在制备cofs过程中,通过构筑模块对cofs的形态、尺寸、表面性质进行调控,从而实现cofs材料的多功能应用,一直是多孔有机高分子领域中的研究热点。三聚喹唑啉是一种具有共轭结构的平面分子,其含有丰富的n原子,具有较强的电负性,能够与温室气体(如co2和ch4)形成较强的相互作用,还能够实现电子在其表面的快速迁移。
技术实现要素:
本发明的目的,是要提供一种以三聚喹唑啉为结点的二维共价有机框架材料,既能用于吸附温室气体、也能用于电催化析氢;
本发明的另一个目的,是要提供上述以三聚喹唑啉为结点的二维共价有机框架材料的制备方法;
本发明的还有一个目的,是要提供上述以三聚喹唑啉为结点的二维共价有机框架材料的应用。
为了实现上述目的,本发明采用的技术方案是:
一种以三聚喹唑啉为结点的二维共价有机框架材料,其化学结构式如下:
本发明还提供了上述以三聚喹唑啉为结点的二维共价有机框架材料的一种制备方法,是在非活性气体保护下,取1,4-二氨基-2,5-二氰基苯通过无水氯化锌催化作用,经离子热合成即得。
作为一种限定,所述离子热合成结束后,还需使用无机酸水溶液、醇类溶剂分别进行洗涤。
作为进一步限定,1,4-二氨基-2,5-二氰基苯与无水氯化锌的摩尔比为1:0.5~2;
所述离子热合成时的反应温度是自室温升温至350~450℃,升温的速率为5℃/min,反应时间为36~72h;
无机酸水溶液为盐酸水溶液、硝酸水溶液或硫酸水溶液;
醇类溶剂为甲醇、乙醇或异丙醇;
无机酸水溶液中的氢离子浓度为0.5~2mol/l。
作为另一种限定,1,4-二氨基-2,5-二氰基苯的制备过程包括依次进行的以下步骤:
a1)在非活性气体保护下,取(±)-2,2'-双(二苯基瞵)-1,1'-联萘(rac-binap)、三(二亚苄-base丙酮)二钯(pd2(dba)3)溶于有机溶剂,催化剂与配体结合后,降至室温再加入2,5-二氯对苯二腈、二苯甲酮亚胺(ph2=nh)和叔丁醇钠,回流反应,然后经分离纯化,得2,5-双((二苯基亚甲基)氨基)对苯二甲腈;
a2)取2,5-双((二苯基亚甲基)氨基)对苯二甲腈溶于四氢呋喃中,加入盐酸水溶液,进行还原反应后,分离纯化,得1,4-二氨基-2,5-二氰基苯盐酸盐;
a3)取1,4-二氨基-2,5-二氰基苯盐酸盐加至氢氧化钠水溶液中,进行中和反应后,分离纯化,得所述1,4-二氨基-2,5-二氰基苯;
该制备过程的化学反应式如下:
作为进一步限定,步骤a1)中,所述分离纯化,是回流反应结束后,冷却至室温,经柱层析提纯,得2,5-双((二苯基亚甲基)氨基)对苯二甲腈;
步骤a2)中,所述分离纯化,是还原反应结束后,浓缩除去溶剂制得1,4-二氨基-2,5-二氰基苯盐酸盐粗品,再使用正己烷洗涤,过滤,干燥,得1,4-二氨基-2,5-二氰基苯盐酸盐;
步骤a3)中,所述分离纯化,是中和反应结束后,加入乙酸乙酯萃取,分相,所得乙酸乙酯相经无水硫酸钠干燥,过滤,浓缩除去溶剂,干燥,得所述1,4-二氨基-2,5-二氰基苯。
作为更进一步限定,步骤a1)中,所述柱层析的洗脱剂是体积比为1:0.5~6的乙酸乙酯和石油醚混合液。
作为进一步限定,步骤a1)中,催化剂与配体结合的温度为40~120℃、时间为1.5~3h;
回流反应的时间为36~72h;
步骤a2)中,还原反应的温度为20~50℃、时间为8~20h;
步骤a3)中,中和反应的温度为0~50℃、时间为0.5~3h。
作为进一步限定,步骤a1)中,有机溶剂为甲苯、乙酸乙酯或二氯甲烷;
(±)-2,2'-双(二苯基瞵)-1,1'-联萘与三(二亚苄-base丙酮)二钯的摩尔比为1:0.25~1;
2,5-二氯对苯二腈、二苯甲酮亚胺及叔丁醇钠三者之间的重量比为1:1~4:1~6;
(±)-2,2'-双(二苯基瞵)-1,1'-联萘作为配体,一般添加量按(±)-2,2'-双(二苯基瞵)-1,1'-联萘与2,5-二氯对苯二腈的重量比0.4~0.6:1;
2,5-二氯对苯二腈与有机溶剂的重量体积比为1kg:15~65l;
步骤a2)中,2,5-双((二苯基亚甲基)氨基)对苯二甲腈与四氢呋喃的重量体积比为1kg:20~50l;
盐酸水溶液的浓度为1~2mol/l;
2,5-双((二苯基亚甲基)氨基)对苯二甲腈与盐酸水溶液中盐酸的摩尔比为1:2~4;
步骤a3)中,氢氧化钠水溶液的浓度为0.5~1.2mol/l;
1,4-二氨基-2,5-二氰基苯盐酸盐与氢氧化钠的摩尔比为1:1~4。
本发明也提供了上述以三聚喹唑啉为结点的二维共价有机框架材料的一种应用,所述含氮共轭微孔聚合物网络负载二硫化钼复合材料作为吸附剂用于吸附温室气体(如ch4、co2);或作为催化剂,用于电催化析氢反应中。
由于采用了上述技术方案,本发明与现有技术相比,所取得的技术进步在于:
本发明采用三聚喹唑啉作为结点制备的以三聚喹唑啉为结点的二维共价有机框架材料,具有较大的孔隙结构,其通过化学键(c-n、c-c、c=n)连接,能够显著提高结构的物理化学稳定性。由于以三聚喹唑啉为结点的二维共价有机框架材料具有较大的孔隙率和较强的n原子电负性,能够为co2和ch4等温室气体的吸附提供更多的存储空间,还能够增强与气体分子之间的相互作用力。以三聚喹唑啉为结点的二维共价有机框架材料表面具有丰富的共轭结构,也能够有效促进电子的快速迁移,通过层与层之间范德华力的相互作用形成一维通道,有效促进h+离子的快速传导,使其作为催化剂用于电催析氢反应中,有效提高电催化析氢反应的效率;
本发明所提供的制备方法采用工业原料,生产成本低,绿色无污染;本发明的制备方法简单,操作方便、过程可控,适于工业化生产,所制的以三聚喹唑啉为结点的二维共价有机框架材料作为吸附剂用于吸附温室气体;或作为催化剂,用于电催化析氢反应中制备氢能源。
附图说明
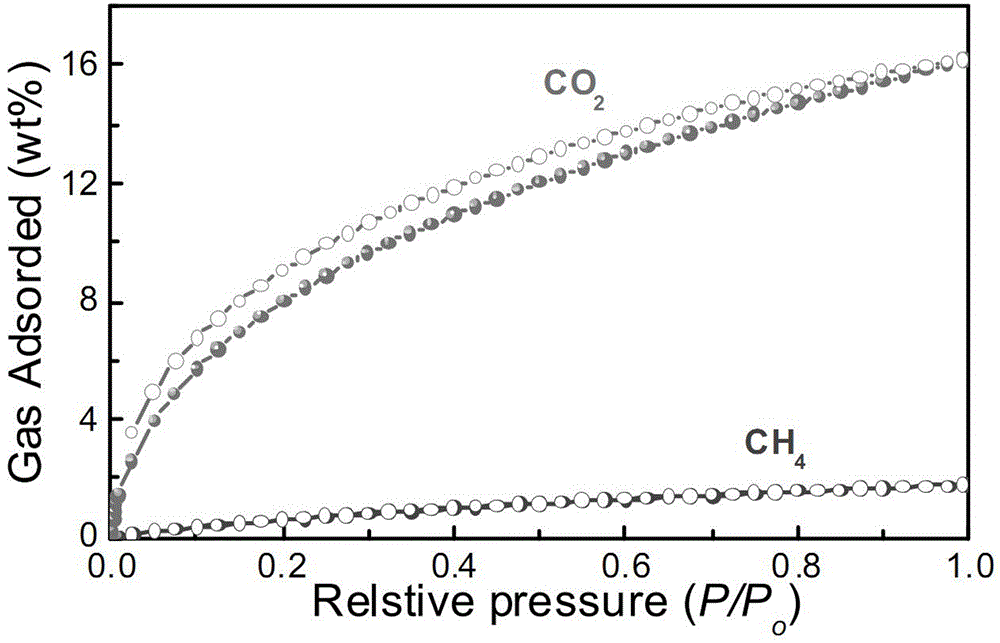
图1是本发明实施例10中测定的实施例1所制备的cofs1的ch4和co2的气体吸附图(273k,1bar);
图2本发明实施例10中测定的实施例1所制备的cofs1和pt20%的伏安线性扫描曲线。
具体实施方式
下面通过具体实施例对本发明做进一步详细说明,应当理解所描述的实施例仅用于解释本发明,并不限定本发明。
下述实施例中所有的试剂如无特殊说明均为现有市售试剂,实验方法如无特殊说明均采用现有的实验方法。
实施例1一种以三聚喹唑啉为结点的二维共价有机框架材料的制备方法
本实施例制备以三聚喹唑啉为结点的二维共价有机框架材料的具体制备过程包括依次进行的以下步骤:
1)制备1,4-二氨基-2,5-二氰基苯
a1)在氮气保护下,取5kg(8mol)的(±)-2,2'-双(二苯基瞵)-1,1'-联萘(rac-binap)、3.66kg(4mol)三(二亚苄-base丙酮)二钯(pd2(dba)3)溶于200l无水甲苯中,形成黑红色的溶液,然后自室温加热升温至110℃搅拌反应2h,使得催化剂与配体结合。待反应结束后冷却至室温,依次加入10kg的2,5-二氯对苯二腈、22.8kg二苯甲酮亚胺和12.7kg叔丁醇钠,加热至回流,回流反应36h,反应结束后,使用硅胶柱层析对反应后溶液进行提纯,洗脱剂是体积比为1:4的乙酸乙酯和石油醚混合液,提纯后蒸去溶剂,60℃真空干燥3h,得19.2kg的2,5-双((二苯基亚甲基)氨基)对苯二甲腈(分子量为486),收率78%。
a2)取6kg的2,5-双((二苯基亚甲基)氨基)对苯二甲腈溶于150l四氢呋喃中,再滴加15l浓度为2mol/l的盐酸水溶液,滴加时长为1h,25℃搅拌进行还原反应8h,反应结束后,浓缩除去溶剂四氢呋喃,依次使用50l正己烷和50l乙醚分别洗涤三次,每次搅拌10min,过滤,60℃真空干燥3h,得2.78kg的1,4-二氨基-2,5-二氰基苯盐酸盐(分子量为231),收率98%;
a3)取2.5kg的1,4-二氨基-2,5-二氰基苯盐酸盐加至50l浓度为0.5mol/l的氢氧化钠水溶液中,25℃搅拌进行中和反应1h,加入25l乙酸乙酯萃取,分相,所得乙酸乙酯相经无水硫酸钠干燥去除多余水分,过滤,浓缩除去溶剂乙酸乙酯,80℃真空干燥12h,得1.6kg的1,4-二氨基-2,5-二氰基苯,收率93%。
制备4-二氨基-2,5-二氰基苯的化学反应式如下:
2)制备以三聚喹唑啉为结点的二维共价有机框架材料
在氮气保护下,取1.58kg(10mol)的1,4-二氨基-2,5-二氰基苯、1.36kg(10mol)无水氯化锌混合后,置于管式炉中以5℃/min的升温速度从室温匀速加热至400℃,并保持400℃反应36h,反应结束后,冷却至室温,研磨产物至粒径500nm,得2.53kg粗产物。将粗产物依次用20l浓度为1mol/l的盐酸水溶液、20l无水乙醇、20l丙酮分别洗涤三次,120℃真空干燥48h,得1.48kg以三聚喹唑啉为结点的二维共价有机框架材料(简称cofs1)。
本实施例制备的以三聚喹唑啉为结点的二维共价有机框架材料可以作为吸附剂用于吸附温室气体,也可以作为催化剂,用于制备氢能源的电催化析氢反应中。
实施例2~9以三聚喹唑啉为结点的二维共价有机框架材料的制备方法
实施例2~9分别为一种以三聚喹唑啉为结点的二维共价有机框架材料的制备方法,它们的步骤与实施例1基本相同,不同之处仅在于工艺参数的不同,具体详见表1:
表1实施例2~9中各项工艺参数一览表
实施例2~9其它部分的内容,与实施例1相同。
实施例10以三聚喹唑啉为结点的二维共价有机框架材料的性能测试
对比实施例1~4所制备的cofs1~cofs4的晶型性能,可知1,4-二氨基-2,5-二氰基苯和无水氯化锌在摩尔比为1:1时,制备的cofs1的晶型性能最好;
对比实施例1制备的cofs1和实施例5制备的cofs5、实施例7制备的cofs7的晶型性能,对比实施例2制备的cofs2和实施例6制备的cofs6、实施例8制备的cofs8的晶型性能,对比实施例4制备的cofs4和实施例9制备的cofs9的晶型性能,可知离子热合成时反应温度为400℃时,制备的cofs的晶型性能最好;
对实施例1制备的cofs1进行co2和ch4气体吸附的实验,取5g的cofs1样品放入脱气管中,150℃脱气12h后,将盛有脱气后的cofs1样品的脱气管接入康塔气体吸附仪中进行测试,参见图1,在273k(开尔文温度,相当于0℃)、1bar的条件下,co2吸附量为16.2wt%,ch4吸附量为1.77wt%;
使用普林斯顿电化学工作站扫描线性伏安曲线,扫描速率为5mv/s,扫描范围0到-1v,测得实施例1制备的cofs1在电流密度为10时,参见图2,过电势为80mv;
20%的pt/c在电流密度为10时,参见图2,过电势为50mv,由于其过电势小,在催化析氢过程中,能耗较小,因此,现有的催化析氢一般采用20%的pt/c作为催化剂电催化析氢。本发明制备的cofs的过电势接近20%的pt/c,这种基于三聚喹唑啉为结点的cofs结构,不仅能够提高cofs在酸性条件下的稳定性,由其组成的全共轭平面还能有效促进电子的离域效应,提高材料的导电性。用于电催化析氢时,本发明制备的cofs能够使得电催化反应能在更低的驱动电流下触发析氢反应,从而降低析氢能耗,同时还能够实现氢的快速吸附和释放,以获得更高的析氢效率和更高的产氢量,显著提高析氢效果。
对实施例2~9制备的cofs2~cofs9进行co2和ch4气体吸附的实验,在273k(开尔文温度,相当于0℃)、1bar的条件下,co2吸附量和ch4吸附量见表2;对实施例2~9制备的cofs2~cofs9进行扫描线性伏安曲线,由于实施例2~9和实施例1测的扫描线性伏安曲线几乎相同,图2中无法看出区别,因此,图2中未显示。在电流密度为10时,实施例2~9制备的cofs2~cofs9的过电势的值见表2:
表2实施例2~9中各项测定结果一览表
需要注意,实施例1~9,仅是本发明的较佳实施例,并非是对本发明所作的其他形式的限定,任何熟悉本专业的技术人员都可能利用上述技术内容作为启示加以变更或改型为等同变化的等效实施例,但凡是未脱离本发明权利要求的技术实质,对以上实施例所作出的简单修改、等同变化与改型,仍属于本发明权利要求保护的范围。
- 还没有人留言评论。精彩留言会获得点赞!