弯曲芳香族化合物及其制备方法与流程
本发明涉及弯曲芳香族化合物及其制备方法。
背景技术:
碳纳米材料是由具有纳米尺寸结构的碳组成的物质的总称,到目前为止,已经发现和研究了富勒烯(球形),碳纳米管(圆柱形)和石墨烯(片状)。由于这些化合物具有优于硅及金属等的常规材料的电气特性及物理性能,因此它们有望用作下一代电子设备的材料。
然而,例如,纳米石墨烯容易在有机溶剂中聚集,并且其低溶解度一直是个问题。
为了解决该问题,伊丹等人开发了由c80h30表示的翘曲纳米石墨烯(wng80)(非专利文献1)。由于wng80具有弯曲结构,因此与平面状石墨烯相比,它在有机溶剂中的溶解性极佳,并且还具有与等平面片状石墨烯不同的特性,所述等平面片状石墨烯可以反复吸收和释放电子并发出绿色荧光。因此,可以期望将其应用于生物成像及有机半导体材料等。
作为赋予该wng80新功能的方法,可以导入取代基或合成具有相似结构的化合物。到目前为止,已经开发了在其中导入了水溶性取代基的水溶性衍生物,并且该化合物可以用作基质辅助激光解吸质谱分析用基质或荧光染料(专利文献1和非专利文献2)。尽管这些wng80在有机溶剂中的溶解性优于纳米石墨烯,但是要应用于有机半导体材料等,还需要在有机溶剂中的进一步的溶解性。
现有技术文献
专利文献
专利文献1:特开2018-154577
非专利文献
非专利文献1:nat.chem.,2013,5,739-744
非专利文献2:angew.chem.int.ed.,2018,57,2874-28787
技术实现要素:
技术问题
本发明的目的在于,提供具有与wng80类的弯曲结构,并且在有机溶剂中显示出比wng80优异的溶解性的化合物、及其制备方法。
技术方案
作为各种研究的结果,本发明的发明人发现了联苯单元比wng80少,由以下通式(1)表示,
式中,
r彼此独立地相同或不同,为选自由卤素原子、硝基、氰基、三氟甲基、羟基、硫醇基、氨基、二苯氨基、咔唑基、碳数为1~20的直链状或支链状的烷基、烷基氨基以及烷氧基组成的组中的取代基,
n分别彼此独立地为0~3的整数,
翘曲纳米石墨烯c68h28(wng68)化合物在特定波长下具有荧光特性,并且显示出比wng80优异的溶解性。
本发明的要旨如下。
1.本发明提供如下的wng68化合物:由以下通式(1)表示,
式中,
r彼此独立地相同或不同,为选自由卤素原子、硝基、氰基、三氟甲基、羟基、硫醇基、氨基、二苯氨基、咔唑基、碳数为1~20的直链状或支链状的烷基、烷基氨基以及烷氧基组成的组中的取代基,
n分别彼此独立地为0~3的整数。
2.本发明提供如下的化合物:由以下通式(2)表示,
式中,x为卤素原子。
3.本发明提供如下的化合物:由以下通式(3)表示,
式中,
r彼此独立地相同或不同,为选自由卤素原子、硝基、氰基、三氟甲基、羟基、硫醇基、氨基、二苯氨基、咔唑基、碳数为1~20的直链状或支链状的烷基、烷基氨基以及烷氧基组成的组中的取代基,
n分别彼此独立地为0~3的整数,
x为卤素原子。
4.本发明提供如下的卤代wng68化合物:由以下通式(4)表示,
式中,
r彼此独立地相同或不同,为选自由卤素原子、硝基、氰基、三氟甲基、羟基、硫醇基、氨基、二苯氨基、咔唑基、碳数为1~20的直链状或支链状的烷基、烷基氨基以及烷氧基组成的组中的取代基,
n分别彼此独立地为0~3的整数,
x为卤素原子。
5.本发明提供通过4所述的化合物的x基的去除反应来制备1所述的化合物的制备方法。
6.本发明提供通过以下化合物(a)与卤化试剂的反应来制备2所述的化合物的制备方法,
7.本发明提供通过2所述的化合物与由以下通式(5)表示的化合物的铃木-宫浦偶联反应来制备3所述的化合物的制备方法。
式中,
r彼此独立地相同或不同,为选自由卤素原子、硝基、氰基、三氟甲基、羟基、硫醇基、氨基、二苯氨基、咔唑基、碳数为1~20的直链状或支链状的烷基、烷基氨基以及烷氧基组成的组中的一种或两种或以上的取代基,
n分别彼此独立地为0~3的整数,
y为离去基。
8.本发明提供通过3所述的化合物的缩合反应制备4所述的化合物的制备方法。
9.本发明提供如下的由以下通式(1)表示的化合物的制备方法,
式中,
r彼此独立地相同或不同,为选自由卤素原子、硝基、氰基、三氟甲基、羟基、硫醇基、氨基、二苯氨基、咔唑基、碳数为1~20的直链状或支链状的烷基、烷基氨基以及烷氧基组成的组中的取代基,
n分别彼此独立地为0~3的整数,
x为卤素原子,
其特征在于,
包括:
获得由以下通式(2)表示的化合物的步骤;
通过由上述通式(2)表示的化合物与由以下通式(5)表示的化合物的铃木-宫浦偶联反应来制备由以下通式(3)表示的化合物的步骤,
通过由上述通式(3)表示的化合物的缩合反应来制备由以下通式(4)表示的化合物的步骤,
发明的效果
根据本发明,可以提供在有机溶剂中显示出比wng80优异的溶解性,并且具有相同堆积结构的化合物。
并且,本发明的发明人发现了将化合物(a)的硼酸酯基的一处选择性的转化为卤素的新方法。
通过该方法可以合成指定了取代位置的wng68前体。因此,可以有效地提供wng68,而无需精切地分离纯化。
并且,wng68化合物及由以下通式(4)表示的卤代wng68化合物与wng80化合物相同地显示出荧光,可以期望将其应用于生物成像及有机半导体材料等,上述由通式(4)表示的卤代wng68化合物如下:
式中,
r彼此独立地相同或不同,为选自由卤素原子、硝基、氰基、三氟甲基、羟基、硫醇基、氨基、二苯氨基、咔唑基、碳数为1~20的直链状或支链状的烷基、烷基氨基以及烷氧基组成的组中的取代基,
n分别彼此独立地为0~3的整数,
x为卤素原子。
此外,在wng68化合物的合成过程中的缩合反应中,抑制了翘曲纳米石墨烯化合物的合成过程中的缩合反应中发生的副反应,并且提高了产率。
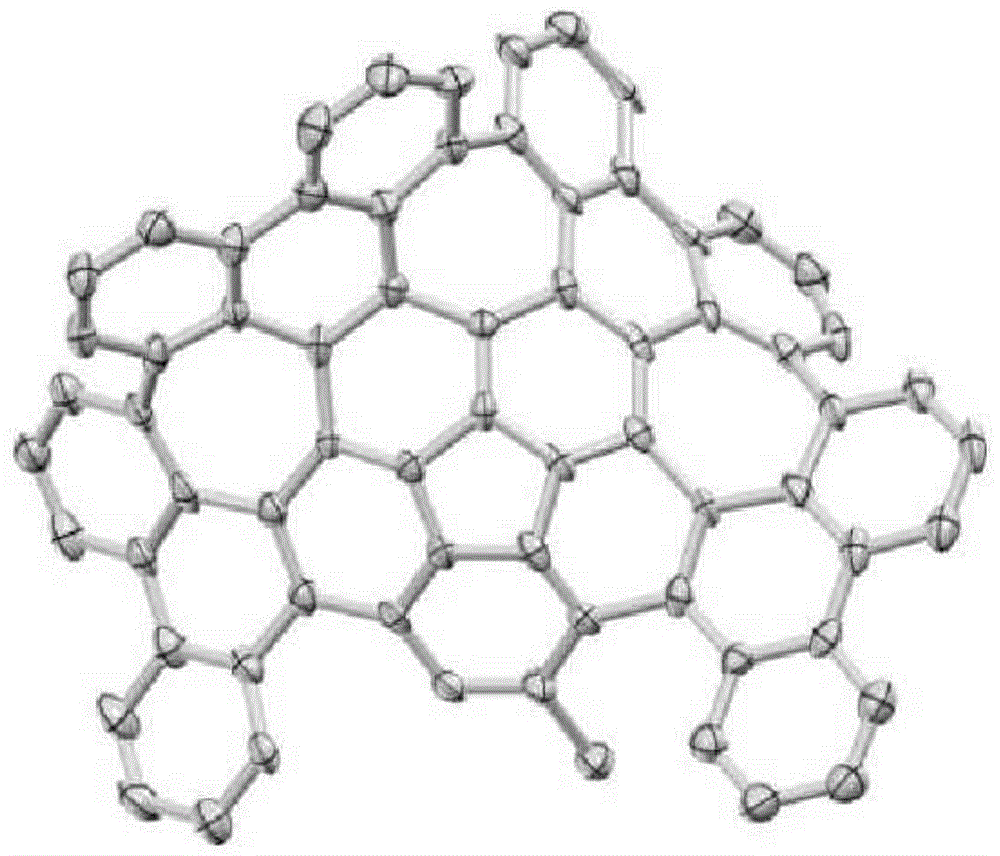
此外,通过x射线晶体结构分析确定了氯化wng68的分子结构。氯化wng68是新型化合物,可以通过交叉偶联反应等方法将分子中的氯基团转化为其他官能团,因此可以赋予新的功能和特性。
附图说明
图1为示出氯化wng68的x射线结构衍射的图。
图2为示出氯化wng68的x射线结构衍射的图。
图3为示出氯化wng68的x射线结构衍射的图。
图4为示出wng68(上段)及氯化wng68(cl-wng68;下段)的吸收光谱(实线)以及荧光光谱(虚线)的图。
具体实施方式
以下,详细说明本发明。
本发明涉及wng68化合物,上述wng68化合物由以下通式(1)表示,
式中,
r彼此独立地相同或不同,为选自由卤素原子、硝基、氰基、三氟甲基、羟基、硫醇基、氨基、二苯氨基、咔唑基、碳数为1~20(如碳数为1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20)的直链状或支链状的烷基、烷基氨基以及烷氧基组成的组中的取代基,
n分别彼此独立地为0~3的整数(如0、1、2或3)。
作为通式(1)中的r的直链状的烷基包括甲基、乙基、丙基、丁基、戊基、己基、庚基、辛基、壬基、癸基、十一烷基、十二烷基、十三烷基、十四烷基、十五烷基、十六烷基、十七烷基、十七烷基。
作为通式(1)中的r的支链状的烷基包括1-甲基乙基、1-甲基丙基、叔丁基、叔戊基、1-乙基丙基、1-正丙基丙基、1-甲基丁基、1-乙基丁基、1-丙基丁基、1-正丁基丁基、1-甲基戊基、1-乙基戊基、1-正丙基戊基、1-正戊基戊基、1-甲基己基、1-乙基己基、1-正丙基己基、1-正丁基己基、1-正戊基己基、1-正己基己基、1-甲基庚基、1-乙基庚基、1-正丙基庚基、1-正丁基庚基、1-正戊基庚基。
从产率及合成的容易性等的观点来看,上述烷基的碳数优选为1~20,更优选为1~12,进一步优选为1~5。
通式(1)中的r的二苯氨基及咔唑基分别可以被取代或未被取代。被取代时的取代基包括烷基、烷氧基、芳基、芳氧基、氨基等。
从合成的容易性的观点来看,通式(1)中的r优选为甲基、乙基、丙基、丁基、戊基、己基、叔丁基、叔戊基,更优选为甲基、乙基、叔丁基,进一步优选为甲基、叔丁基。
通式(1)中的n优选为0或1,更优选为0。
本发明还涉及由以下通式(2)表示的化合物,
式中,x为卤素原子。
卤素原子x优选为氟原子、氯原子、溴原子、以及碘原子,更优选为氯原子、溴原子、以及碘原子,进一步优选为氯原子。
本发明还涉及由以下通式(3)表示的化合物,
式中,
r彼此独立地相同或不同,为选自由卤素原子、硝基、氰基、三氟甲基、羟基、硫醇基、氨基、二苯氨基、咔唑基、碳数为1~20(如碳数为1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20)的直链状或支链状的烷基、烷基氨基以及烷氧基组成的组中的取代基,
n分别彼此独立地为0~3(如0、1、2或3)的整数,
x为卤素原子。
作为通式(3)中的r的直链状的烷基包括甲基、乙基、丙基、丁基、戊基、己基、庚基、辛基、壬基、癸基、十一烷基、十二烷基、十三烷基、十四烷基、十五烷基、十六烷基、十七烷基、十七烷基。
作为通式(3)中的r的支链状的烷基包括1-甲基乙基、1-甲基丙基、叔丁基、叔戊基、1-乙基丙基、1-正丙基丙基、1-甲基丁基、1-乙基丁基、1-丙基丁基、1-正丁基丁基、1-甲基戊基、1-乙基戊基、1-正丙基戊基、1-正戊基戊基、1-甲基己基、1-乙基己基、1-正丙基己基、1-正丁基己基、1-正戊基己基、1-正己基己基、1-甲基庚基、1-乙基庚基、1-正丙基庚基、1-正丁基庚基、1-正戊基庚基。
从产率及合成的容易性等的观点来看,上述烷基的碳数优选为1~20,更优选为1~12,进一步优选为1~5。
通式(3)中的r的二苯氨基及咔唑基分别可以被取代或未被取代。被取代时的取代基包括烷基、烷氧基、芳基、芳氧基、氨基等。
从合成的容易性的观点来看,通式(3)中的r优选为甲基、乙基、丙基、丁基、戊基、己基、叔丁基、叔戊基,更优选为甲基、乙基、叔丁基,进一步优选为甲基、叔丁基。
通式(3)中的n优选为0或1,更优选为0。
通式(3)中的卤素原子x优选为氟原子、氯原子、溴原子、以及碘原子,更优选为氯原子、溴原子、以及碘原子,进一步优选为氯原子。
本发明还涉及由以下通式(4)表示的化合物,
式中,
r彼此独立地相同或不同,为选自由卤素原子、硝基、氰基、三氟甲基、羟基、硫醇基、氨基、二苯氨基、咔唑基、碳数为1~20(如碳数为1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20)的直链状或支链状的烷基、烷基氨基以及烷氧基组成的组中的取代基,
n分别彼此独立地为0~3的整数(如0、1、2或3),
x为卤素原子。
作为通式(4)中的r的直链状的烷基包括甲基、乙基、丙基、丁基、戊基、己基、庚基、辛基、壬基、癸基、十一烷基、十二烷基、十三烷基、十四烷基、十五烷基、十六烷基、十七烷基、十七烷基。
作为通式(4)中的r的支链状的烷基包括1-甲基乙基、1-甲基丙基、叔丁基、叔戊基、1-乙基丙基、1-正丙基丙基、1-甲基丁基、1-乙基丁基、1-丙基丁基、1-正丁基丁基、1-甲基戊基、1-乙基戊基、1-正丙基戊基、1-正戊基戊基、1-甲基己基、1-乙基己基、1-正丙基己基、1-正丁基己基、1-正戊基己基、1-正己基己基、1-甲基庚基、1-乙基庚基、1-正丙基庚基、1-正丁基庚基、1-正戊基庚基。
从产率及合成的容易性等的观点来看,上述烷基的碳数优选为1~20,更优选为1~12,进一步优选为1~5。
通式(4)中的r的二苯氨基及咔唑基分别可以被取代或未被取代。被取代时的取代基包括烷基、烷氧基、芳基、芳氧基、氨基等。
从合成的容易性的观点来看,通式(4)中的r优选为甲基、乙基、丙基、丁基、戊基、己基、叔丁基、叔戊基,更优选为甲基、乙基、丙基、叔丁基,进一步优选为甲基、叔丁基。
通式(4)中的卤素原子x优选为氟原子、氯原子、溴原子、以及碘原子,更优选为氯原子、溴原子、以及碘原子,进一步优选为氯原子。
本发明还涉及通过由以下通式(4)表示的化合物的x基的去除反应来制备由通式(1)表示的化合物的制备方法,
式中,
r彼此独立地相同或不同,为选自由卤素原子、硝基、氰基、三氟甲基、羟基、硫醇基、氨基、二苯氨基、咔唑基、碳数为1~20(如碳数为1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20)的直链状或支链状的烷基、烷基氨基以及烷氧基组成的组中的取代基,
n分别彼此独立地为0~3的整数(如0、1、2或3),
x为卤素原子。
本方法中的优选x与对由通式(4)表示的化合物的x所述的优选实施方式相同。
本方法中的优选r与对由通式(4)表示的化合物的r所述的优选实施方式相同。
x基的去除反应通过金属锂、金属钠、金属钾等的单金属、三乙基硅烷、氢化铝锂、氢化三氯甲烷锂等的还原剂、以及使用钯催化剂和甲酸的还原反应来进行,其优选通过使用钯催化剂及甲酸的还原反应或金属锂、金属钠来进行,更优选通过金属锂来进行。通常,相对于1摩尔的由通式(4)表示的化合物,上述还原剂优选为1摩尔~50摩尔,更优选为5摩尔~30摩尔。
当通过单金属去除时,可以任意添加电子转移剂。电子转移剂包括未取代的萘、n,n-二甲基-1-萘胺等的取代萘、取代或未取代的联苯化合物等,优选为未取代的萘。
当进行使用钯催化剂及甲酸的还原反应时,对钯催化剂没有特别限制,除了金属钯以外还包括作为有机化合物等的合成用催化剂公知的钯化合物等。具体地,作为钯催化剂包括四(三苯基膦)钯、三(二亚苄基丙酮)二钯(pd2(dba)3)、双(二亚苄基丙酮)钯、双(三叔丁基膦基)钯、乙酸钯、卤化钯(pdcl2、pdbr2、pdi2)等中的一种或两种或以上。在本发明中,从反应产率等的观点来看,优选为乙酸钯。通常,相对于1摩尔的由通式(4)表示的化合物,钯催化剂的使用量优选为0.01摩尔~1.00摩尔,更优选为0.5摩尔~1.0摩尔。
在使用钯催化剂及甲酸的还原反应中,需要使用甲酸。通常,相对于1摩尔的由通式(4)表示的化合物,甲酸的使用量优选为2摩尔~30摩尔,更优选为10摩尔~20摩尔。
在使用钯催化剂及甲酸的还原反应中,可以与上述钯催化剂一起使用可以与钯原子配位的配体化合物。尽管不使用配体化合物也可以进行反应,但是通过使用配体化合物可以进一步提高反应产率。这种配体化合物优选为膦化合物,包括三苯基膦、三三甲氧基膦、三甲氧基膦、三乙膦、三异丙基膦、三异丙氧基膦、三环戊基膦、三苯甲基膦、二苯基膦基甲烷、1,2-双(二苯基膦基)乙烷、2-二环己基膦基2’,6’-二甲氧基联苯(sphos)等的一种或两种或以上。这种配体化合物可以是溶剂化物。从反应产率等的观点来看,优选为2-二环己基膦基2’,6’-二甲氧基联苯(sphos)。从反应产率等的观点来看,相对于1摩尔的钯催化剂,配体化合物的使用量优选为0.5摩尔~10.0摩尔,更优选为1.0摩尔~3.0摩尔。
在使用钯催化剂及甲酸的还原反应中,优选使用碱。作为碱,优选烷基胺。作为这种碱包括三甲胺、三乙胺、二异丙基乙胺、三丁胺等的一种或两种或以上。其中,在本步骤中,从合成的容易性、产率等的观点来看,优选烷基胺,更优选三乙胺。在本发明中,从合成的容易性、产率等的观点来看,相对于甲酸,碱的使用量优选过量,通常,相对于使用1摩尔的甲酸,碱的使用量优选为1.1摩尔~3.0摩尔,更优选为1.5摩尔~2.0摩尔。
使用单金属的x基的去除反应优选在惰性气体气氛(氮气、氩气等)下进行,通常可以在溶剂中进行。作为溶剂包括二乙醚、二异丙醚、二丁醚、二甲氧基乙烷、环戊基甲基醚、叔丁基甲基醚等的链状醚;四氢呋喃(thf)、二恶烷等的环状醚;戊烷、己烷、庚烷、环己烷等的脂族烃;二氯甲烷、二氯乙烷、氯仿、四氯化碳等的脂族卤代烃;苯、甲苯、二甲苯、均三甲苯等的芳族烃;氯苯、三氟甲苯等的芳香卤代烃;以及甲醇、乙醇、异丙醇等的醇等的一种或两种或以上。在本步骤中,从产率和合成的容易性的观点来看,优选为四氢呋喃(thf)、二恶烷等的环状醚,更优选为四氢呋喃(thf)。
通过使用钯催化剂及甲酸的还原反应的x基的去除反应优选在惰性气体气氛(氮气、氩气等)下进行,通常可以在溶剂中进行。作为溶剂包括:二乙醚、二异丙醚、二丁醚、二甲氧基乙烷、环戊基甲基醚、叔丁基甲基醚等的链状醚;四氢呋喃(thf)、二恶烷等的环状醚;戊烷、己烷、庚烷、环己烷等的脂族烃;二氯甲烷、二氯乙烷、氯仿、四氯化碳等的脂族卤代烃;苯、甲苯、二甲苯、均三甲苯等的芳族烃;氯苯、三氟甲苯等的芳香卤代烃;甲醇、乙醇、异丙醇等的醇;以及n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、n-甲基吡咯烷酮等的酰胺的一种或两种或以上。在本步骤中,从产率和合成的容易性的观点来看,优选为n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、n-甲基吡咯烷酮等的酰胺,更优选为n,n-二甲基甲酰胺。
使用单金属的x基的去除反应可以在加热下、常温下、以及冷却下进行,通常,优选-78℃~100℃左右,更优选为-78℃~50℃左右,进一步优选为0℃。反应时间可以是反应进行的时间,通常,优选为10分钟~3天左右,更优选为30分钟~1天左右。
通过使用钯催化剂及甲酸的还原反应的x基的去除反应可以在加热下、常温下、以及冷却下进行,通常,优选为-78℃~150℃左右,更优选为室温~150℃左右,进一步优选为50℃~100℃。反应时间可以是反应进行的时间,通常,优选为1小时~1天左右,更优选为2小时~4小时左右。
反应完成后,可以根据需要进行通过硅藻土或硅胶等的过滤、液体分离操作等的常规的后处理和硅胶柱色谱法、重结晶、沉淀纯化等的纯化步骤。
本发明还涉及通过化合物(a)与卤化试剂的反应来制备由以下通式(2)表示的化合物,
式中,x为卤素原子。
本方法中的优选x与对由通式(2)表示的化合物所述的优选实施方式相同。
卤化试剂没有特别限制,但包括氟化铜(i)、氯、n-氯琥珀酰亚胺、氯化铜(i)、n-氯糖精、三氯异氰尿酸、1,3-二氯-5,5-二甲基海因、溴、n-溴代琥珀酰亚胺、溴化铜(i)、n-溴糖精、二溴异氰尿酸、1,3-二溴-5,5-二甲基海因、碘、n-碘代琥珀酰亚胺、碘化铜(i)、n-碘糖精等。从产率和合成的容易性等的观点来看,优选为n-氯糖精、n-溴糖精、n-碘糖精,更优选为n-氯糖精。
与卤化试剂的反应通常可以在溶剂中进行。例如,二乙醚、二异丙醚、二丁醚、二甲氧基乙烷、环戊基甲基醚、叔丁基甲基醚等的链状醚;四氢呋喃(thf)、二恶烷等的环状醚;戊烷、己烷、庚烷、环己烷等的脂族烃;二氯甲烷、二氯乙烷、氯仿、四氯化碳等的脂族卤代烃;苯、甲苯、二甲苯、均三甲苯等的芳族烃;氯苯、三氟甲苯等的芳香卤代烃;以及甲醇、乙醇、异丙醇等的醇等的一种或两种或以上。从产率和合成的容易性的观点来看,优选为二乙醚、二异丙醚、二丁醚、二甲氧基乙烷、环戊基甲基醚、叔丁基甲基醚等的链状醚;以及四氢呋喃(thf)、二恶烷等的环状醚、二氯甲烷、二氯乙烷、氯仿、四氯化碳等的脂族卤代烃,更优选为二氯甲烷、二氯乙烷、氯仿、四氯化碳等的脂族卤代烃,最优选为二氯甲烷。
与卤化试剂的反应可以在惰性气体气氛(氮气、氩气等)下或在大气中进行。
与卤化试剂的反应可以在反应温度为加热下、常温下以及冷却下进行,通常,优选为0℃~100℃左右,更优选为0℃~50℃左右,进一步优选为室温(25℃)。反应时间可以是反应进行的时间,通常,优选为10分钟~3天左右,更优选为30分钟~1天左右。
反应完成后,可以根据需要进行通过硅藻土或硅胶等的过滤、液体分离操作等的常规的后处理和硅胶柱色谱法、重结晶、沉淀纯化等的纯化步骤。
本发明还涉及通过由以下通式(2)表示的化合物与由以下通式(5)表示的化合物的铃木-宫浦偶联反应来制备由以下通式(3)表示的化合物的制备方法,
式中,
r彼此独立地相同或不同,为选自由卤素原子、硝基、氰基、三氟甲基、羟基、硫醇基、氨基、二苯氨基、咔唑基、碳数为1~20(如为1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20)的直链状或支链状的烷基、烷基氨基以及烷氧基组成的组中的一种或两种或以上的取代基,
n分别彼此独立地为0~3的整数(如0、1、2或3),
x为卤素原子,
y为离去基。
本方法中的优选r、n、以及x分别与对由通式(3)表示的化合物所述的优选实施方式相同。
从产率及合成的容易性等的观点来看,通式(5)中的离去基y优选为-otf以及卤素,更优选为-otf、溴、以及碘,进一步优选为碘。
相对于由通式(3)表示的化合物,铃木-宫浦偶联反应中使用的由通式(5)表示的化合物的量优选过量,通常,相对于1摩尔的由通式(3)表示的化合物,优选为2摩尔~20摩尔,更优选为5摩尔~10摩尔。
作为铃木-宫浦偶联反应中的钯催化剂没有特别限制,除了金属钯以外还包括作为有机化合物等的合成用催化剂公知的钯化合物等。具体地,作为钯催化剂包括四(三苯基膦)钯、三(二亚苄基丙酮)二钯(pd2(dba)3)、双(二亚苄基丙酮)钯、双(三叔丁基膦基)钯、乙酸钯、卤化钯(pdcl2、pdbr2、pdi2)等中的一种或两种或以上。在本发明中,从反应产率等的观点来看,优选为三(二亚苄基丙酮)二钯(pd2(dba)3)。通常,相对于1摩尔的由通式(4)表示的化合物,钯催化剂的使用量优选为0.01摩尔~1.00摩尔,更优选为0.02摩尔~0.50摩尔。
在铃木-宫浦偶联反应中,可以与上述钯催化剂一起使用可以与钯原子配位的配体化合物。尽管不使用配体化合物也可以进行反应,但是通过使用配体化合物可以进一步提高反应产率。这种配体化合物优选为膦化合物,包括三苯基膦、三三甲氧基膦、三甲氧基膦、三乙膦、三异丙基膦、三异丙氧基膦、三环戊基膦、三苯甲基膦、二苯基膦基甲烷、1,2-双(二苯基膦基)乙烷、2-二环己基膦基2’,6’-二甲氧基联苯(sphos)等的一种或两种或以上。这种配体化合物可以是溶剂化物。从反应产率等的观点来看,优选为三(4-甲氧基苯基)膦。从反应产率等的观点来看,相对于1摩尔的钯催化剂,配体化合物的使用量优选为0.5摩尔~10.0摩尔,更优选为1.0摩尔~5.0摩尔。
在铃木-宫浦偶联反应中,优选使用碱。作为碱,优选使用碱金属磷酸盐、碱金属碳酸盐、碱金属氟化物盐等。作为这种碱包括:磷酸锂、磷酸钠、磷酸钾等的碱金属磷酸盐;碳酸锂、碳酸钠、碳酸钾、碳酸铯等的碱金属碳酸盐;以及氟化钠、氟化钾、氟化铯等的碱金属氟化物盐等的一种或两种或以上。其中,从合成的容易性、产率等的观点来看,在本步骤中,优选为碱金属磷酸盐,更优选为磷酸钾。在本发明中,从合成的容易性、产率等的观点来看,相对于由通式(3)表示的化合物,碱的使用量优选过量,通常,优选为2摩尔~20摩尔的由通式(3)表示的化合物,更优选为5摩尔~10摩尔。
上述反应通常可以在溶剂中进行。作为溶剂,例如,除了水还可以包括:二乙醚、二异丙醚、二丁醚、二甲氧基乙烷、环戊基甲基醚、叔丁基甲基醚等的链状醚;四氢呋喃(thf)、二恶烷等的环状醚;戊烷、己烷、庚烷、环己烷等的脂族烃;二氯甲烷、二氯乙烷、氯仿、四氯化碳等的脂族卤代烃;苯、甲苯、二甲苯、均三甲苯等的芳族烃;氯苯、三氟甲苯等的芳香卤代烃;以及甲醇、乙醇、异丙醇等的醇等的一种或两种或以上。在本步骤中,从产率和合成的容易性的观点来看,优选为水以及芳族烃,更优选为水和甲苯的组合。
上述反应优选在惰性气体气氛(氮气、氩气等)下进行,反应温度可以是加热下、常温下、冷却下中的任何一种,通常,优选为室温(25℃)~150℃左右,更优选为50℃~100℃左右。反应时间可以是反应进行的时间,通常,优选为10分钟~48小时左右,更优选为30分钟~36小时左右。反应完成后,可以根据需要进行通过硅藻土或硅胶等的过滤、液体分离操作等的常规的后处理和硅胶柱色谱法、重结晶、沉淀纯化等的纯化步骤。
本发明还涉及通过由以下通式(3)表示的化合物的缩合反应来制备由以下通式(4)表示的化合物的制备方法,
式中
r彼此独立地相同或不同,为选自由卤素原子、硝基、氰基、三氟甲基、羟基、硫醇基、氨基、二苯氨基、咔唑基、碳数为1~20(如为1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20)的直链状或支链状的烷基、烷基氨基以及烷氧基组成的组中的取代基,
n分别彼此独立地为0~3的整数(如0、1、2或3),
x为卤素原子。
本方法中的优选r、n、以及x分别与对由通式(4)表示的化合物所述的优选实施方式相同。
缩合反应通常可以在溶剂中进行。作为溶剂包括:二乙醚、二异丙醚、二丁醚、二甲氧基乙烷、环戊基甲基醚、叔丁基甲基醚等的链状醚;四氢呋喃(thf)、二恶烷等的环状醚;戊烷、己烷、庚烷、环己烷等的脂族烃;二氯甲烷、二氯乙烷、氯仿、四氯化碳等的脂族卤代烃;苯、甲苯、二甲苯、均三甲苯等的芳族烃;氯苯、三氟甲苯等的芳香卤代烃;以及甲醇、乙醇、异丙醇等的醇等的一种或两种或以上。从产率和合成的容易性的观点来看,优选为脂族卤代烃,更优选为二氯甲烷。
用于进行缩合反应的氧化剂包括邻氯苯醌、对氯苯醌、氯化铁(iii)、2,3-二氯-5,6-二氰基对苯醌(ddq)、二氧化锰、琼斯试剂、氯铬酸吡啶鎓、重铬酸吡啶鎓、硝酸铈铵(can)、碘等的一种或两种或以上。在本步骤中,从产率和合成的容易性的观点来看,优选为2,3-二氯-5,6-二氰基对苯醌(ddq)。通常,相对于由通式(3)表示的化合物,氧化剂的使用量优选过量,例如,相对于1摩尔的由通式(3)表示的化合物,优选为5摩尔~20摩尔,更优选为8摩尔~15摩尔。
在缩合反应中,可以任意使用添加剂。这种添加剂包括甲磺酸、甲苯磺酸、三氟甲磺酸等,优选为三氟甲磺酸。
添加剂的使用量没有特别限制。从产率等的观点来看,相对于由通式(3)表示的化合物,优选过量。优选使用10摩尔或以上,更优选使用100摩尔,进一步优选使用溶剂量。
缩合反应可以在惰性气体气氛(氮气、氩气等)下或在大气中进行,优选在惰性气体气氛(氮气、氩气等)下进行。缩合反应中的反应温度为加热下、常温下以及冷却下,通常,优选为-78℃~100℃左右,更优选为-78℃~50℃左右,进一步优选0℃。反应时间可以是反应进行的时间,通常,优选为10分钟~3天左右,更优选为30分钟~1天左右。
反应完成后,可以根据需要进行通过硅藻土或硅胶等的过滤、液体分离操作等的常规的后处理和硅胶柱色谱法、重结晶、沉淀纯化等的纯化步骤。
本发明包括由以下通式(1)表示的化合物的制备方法,
式中,
r彼此独立地相同或不同,为选自由卤素原子、硝基、氰基、三氟甲基、羟基、硫醇基、氨基、二苯氨基、咔唑基、碳数为1~20(如为1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20)的直链状或支链状的烷基、烷基氨基以及烷氧基组成的组中的取代基,
n分别彼此独立地为0~3的整数(如0、1、2或3),
x为卤素原子,
其特征在于,
包括:
获得由以下通式(2)表示的化合物的步骤,
通过由上述通式(2)表示的化合物与由以下通式(5)表示的化合物的铃木-宫浦偶联反应来制备由以下通式(3)表示的化合物的步骤,
通过由上述通式(3)表示的化合物的缩合反应来制备由以下通式(4)表示的化合物的步骤,
本方法中的优选r、n、x分别与对由通式(1)表示的化合物所述的优选实施方式相同。
本方法中的优选y与对由通式(5)表示的化合物所述的优选实施方式相同。
实施例
以下,参照本发明的实施例更详细地说明,但这些仅用于例示本发明的特定实例,本发明不限于此。
合成例1:化合物(b)的合成
根据以往报告(j.am.chem.soc.134,15169-15172)合成了化合物(b)。
向化合物(a)(1103mg,1.25mmol,1.0当量)的二氯甲烷(12.5ml)溶液中加入n-氯糖精(712mg,3.27mmol,2.6当量),在室温下,在大气中,搅拌1小时。向上述混合物加入正己烷,通过硅藻土过滤析出不溶物后,在减压下蒸发溶剂。再次加入正己烷,通过硅藻土过滤。浓缩滤液,得到黄色泡沫状化合物(b)(1018mg,quant.)。
1hnmr(600mhz,c2d2cl4,25℃)δ8.99(s,2h),8.95(s,1h),8.59(s,1h),1.49-1.47(m,48h);13cnmr(150mhzc2d2cl4,25℃)δ139.5(ch),138.5(ch),138.3(ch),137.0(4°),136.6(4°),135.7(4°),135.0(4°),134.1(4°),133.7(ch),133.5(4°),133.0(4°),132.5(4°),131.4(4°),129.7(4°),128.1(ch),127.6(4°),83.91(4°),24.90(ch3);hrms(maldi-tofms)m/z计算值c44h53b4clo8[m·]+:788.3801,实测值:788.3804;mp:>300℃。
合成例2:化合物(c)的合成
在氩气气氛下,在反应容器中装入化合物(b)(63mg,0.08mmol,1.0当量)、k3po4(170mg,0.8mmol,10当量)、pd2(dba)3(7.3mg,0.008mmol,10mol%)、三(4-甲氧基苯基)膦(5.6mg,0.016mmol,20mol%)。加入甲苯(1.33ml)、离子交换水(0.67ml)、2-碘联苯(112mg,0.4mmol,5当量),在氩气气氛下,在70℃下,搅拌24小时,进行铃木-宫浦偶联反应。加入离子交换水(5.0ml)以终止反应后,用二氯甲烷萃取3次,用na2so4干燥合并的有机层,在减压下蒸发溶剂。用甲醇和正己烷洗涤所得粗产物,的到黄色固体化合物(c)(44.9mg,自化合物(a)的产率为44%)。
1hnmr(600mhz,c2d2cl4,25℃)δ7.87-6.85(brm,41h);δ142.69(4°),142.04(4°),141.75(4°),141.38(4°),140.83(4°),140.39(4°),137.81(4°),137.44(4°),134.92(4°),134.44(4°),134.24(4°),133.56(4°),133.13(4°),131.24(ch),131.08(ch),130.73(ch),130.50(4°),130.33(ch),130.15(ch),130.04(ch),129.83(ch),129.67(ch),129.35(4°),129.10(4°),128.52(ch),128.16(ch),128.00(ch),127.73(ch),127.38(ch),127.18(ch),126.47(ch),126.31(ch),126.07(ch),125.12(ch),124.06(ch),123.43(4°),120.19(4°);hrms(maldi-tofms)m/z计算值c68h41cl[m·]+:892.2897,实测值:892.2854;mp:180℃-182℃。
合成例3:化合物(d)(氯化wng68)的合成
在氩气气氛下,相对于二氯甲烷(1ml)中的三氟甲磺酸(0.5ml),在0℃下,滴加化合物(c)(45mg,0.05mmol)和ddq(81mg,0.36mmol)的二氯甲烷(8.5ml)溶液。此外,在0℃下,搅拌20小时。20小时后,用三乙胺(1ml)终止反应,加入甲醇,并通过过滤收集析出的沉淀物。通过柱色谱法(展开液:氯仿)纯化得到的沉淀物,得到黄褐色结晶-氯化wng68(化合物(d)、22mg,49%)。
1hnmr(600mhz,c2d2cl4,25℃)δ9.07(d,j=7.8hz,1h),8.76(d,j=7.2hz,1h),8.63-8.61(m,4h),8.55(d,j=7.8hz,1h),8.49(d,j=8.4hz,1h),8.46(bs,1h),8.40-8.38(m,2h),8.29(s,1h),7.92(t,j=7.2hz,2h),7.88-7.82(m,3h),7.72-7.67(m,2h),7.52(t,j=7.8hz,2h),7.42(d,j=8.4hz,1h),7.38(d,j=7.2hz,1h),7.27(bs,1h),7.18(d,j=7.8hz,1h),7.04(bs,1h),6.98(d,j=6.6hz,1h);13cnmr(150mhz,c2d2cl4,25℃)δ140.02(4°),139.53(4°),139.32(4°),138.99(4°),138.76(4°),137.99(4°),137.87(4°),136.19(4°),135.86(4°),134.149(4°),133.86(4°),133.10(ch),132.59(4°),132.55(4°),132.22(4°),132.12(4°),132.07(4°),131.75(ch),131.71(ch),131.68(4°),131.54(ch),131.39(4°),131.33(4°),131.23(ch),131.00(4°),130.86(4°),130.77(4°),130.65(4°),130.49(4°),130.40(4°),130.19(4°),130.13(ch),129.96.(4°),129.53(ch),129.51(4°),129.29(4°),129.13(4°),128.86.(ch),128.67.(4°),128.63(4°),128.60(4°),128.56(4°),128.40(ch),128.35(ch),128.16(4°),127.86(ch),127.73.(ch),127.56(4°),127.17(ch),126.97(4°),126.80(4°),126.76(ch),126.43(4°),125.32(ch),123.59(ch),122.70(ch),122.37(ch),121.74(ch),121.53(ch),121.41(ch),120.54(ch),120.19(4°),无法观察到四碳原子(4°×2,ch×2);hrms(maldi-tofms)m/z计算值c68h27cl[m·]+:878.1801,实测值:878.1776;mp:>300℃。
合成例4:wng68的合成
在氩气气氛下,在0℃下,将萘(47mg,0.37mmol)加入到thf(0.6ml)中的金属锂(2.1mg,0.30mmol)。进而,在室温下,搅拌3小时。3小时后,冷却至0℃,滴加化合物(d)(11mg,0.013mmol)的thf(2.5ml)溶液。此外,在0℃下,搅拌1小时后,加入饱和氯化铵水溶液(3ml)和水(3ml)以终止反应,用二氯甲烷萃取3次,用na2so4干燥合并的有机层,在减压下蒸发溶剂。向残渣加入甲醇,并通过过滤收集产生的沉淀物。用甲苯重结晶获得的粗产物,得到黄色固体化合物wng68(2.7mg,25%)。
1hnmr(600mhz,c2d2cl4,25℃)δ9.30(d,j=7.8hz,1h),8.71(s,1h),8.67(d,j=7.2hz,1h),8.60(d,j=7.2hz,1h),8.57(d,j=8.4hz,2h),7.81(t,j=6.0hz,1h),7.79(t,j=6.6hz,1h),7.72(t,j=7.2hz,1h),7.71(t,j=7.2hz,1h),7.67(t,j=7.8hz,1h),7.46(d,j=7.8hz,1h),7.42(d,j=7.8hz,1h),7.21(d,j=6.6hz,1h);13cnmr(150mhz,c2d2cl4,25℃)δ139.44(4°),139.41(4°),138.82(4°),134.08(4°),134.05(4°),133.08(4°),133.26(4°),132.61(4°),132.55(4°),132.08(ch),131.75(4°),131.02(4°),130.91(4°),130.83(4°),130.80(4°),130.75(ch),130.61(ch),130.43(4°),130.01(4°),129.43(4°),129.08(4°),129.00(ch),128.60(ch),128.01(ch,2c),127.61(ch),127.50(ch),127.43(4°),127.27(4°),126.87(4°),126.70(ch),123.29(ch),121.44(ch),121.39(ch,2c),无法观察到一个碳(4°);hrms(maldi-tofms)m/z计算值c68h28[m·]+:844.2191,实测值:844.2191;mp:>300℃。
试验例1:化合物(d)(氯化wng68)的x射线晶体衍射
对于氯化wng68,将晶体数据的详细信息以及强度数据收集参数的概要示于表1中。并且,将通过x射线结构衍射确定的氯化wng68结构示于图1~3中。
在任意情况下,用矿物油将适当的晶体固定在玻璃纤维上,转移到rigakupilatus衍射仪的测角仪上。使用了石墨单色mokα射线。通过使用(sir-97)(j.appl.crystallogr.32,115-119.)的直接方法确定结构,并使用yadokari-xg程序(晶体结构分析软件(softwareforcrystalstructureanalyses),2001年),通过对于f2(shelxl-2014/3)(actacrystallogr.,sect.a64,112-122.)的全矩阵最小二乘法将其细化。通过洛伦兹和极化效应校正了强度。各向异性地细化了非氢原子。使用afix指令放置了氢原子。
表1
试验例2:化合物(d)(氯化wng68)的吸收以及荧光光谱测定
所有测定均使用各边1cm的正方形石英池中脱气光谱级二氯甲烷中的稀溶液。在shimadzuuv-3510光谱仪上以0.5nm的分辨率记录uv-vis吸收光谱。用f-4500日立光谱仪或shimadzurf-6000以0.4nm的分辨率测定荧光光谱。使用配备校准积分球系统(207-21460-41)的shimadzurf-6000测定绝对荧光量子产率(φf)。
所获得的wng68显示出荧光,并在有机溶剂中显示出比wng80优异的溶解性。
- 还没有人留言评论。精彩留言会获得点赞!