一种异噁唑啉类驱虫药的制备方法与流程
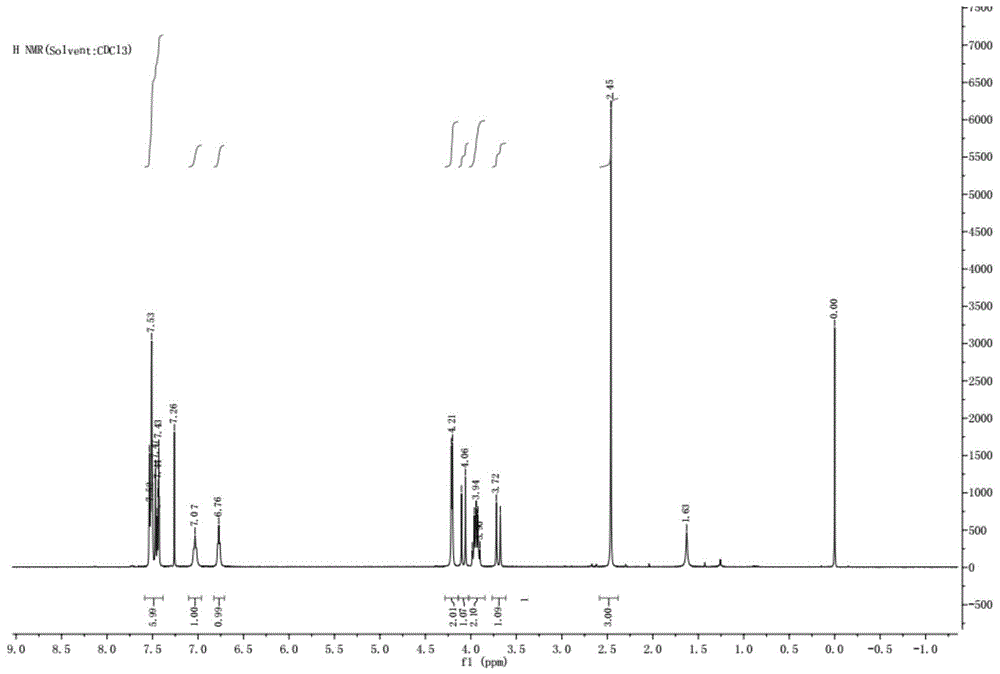
本发明属于化学药物合成
技术领域:
,尤其涉及一种异噁唑啉类驱虫药的制备方法。
背景技术:
:fluralaner(氟雷拉纳),2014年已作为杀虫类兽药(商品名bravecto)在欧美市场销售,主要用于动物体内外杀虫,上市的药物成分为r和s构型的混合物,该化合物cas号:864731-61-3;化学分子式:c22h17cl2f6n3o3,化学名称为4-[5-(3,5-二氯苯基)-4,5-二氢-5-三氟甲基-3-异恶唑基]-2-甲基-氮-[2-氧代-2-[(2,2,2-三氟乙基)氨基]苯甲酰胺,是一类很有效的杀虫活性化合物;fluralaner是最早并且是唯一一种能够快速有效杀灭虱子和跳蚤的兽用医疗用品,对哺乳动物安全、杀虫活性较高、且广谱的新型异恶唑啉类动物杀虫剂。其作为最新一代的兽药,国内并没有相关的学术研究以及商业方面的报道。因此,加强对该类含氟杂环化合物的研究显得尤其重要,可以改善国内动物用药情况,促进国内关于此类药物的开发与研究以及降低药费用具有重大的现实意义。现有技术中提供了一种氟雷拉纳的合成工艺(湖北工业大学,黄道友,新型兽药fluralaner的合成工艺研究,2017),分两步进行,第一步:向烧瓶中加入碘化钾与过硫酸氢钾复合盐,加水溶解,再加入中间体iii和中间体ii,室温搅拌反应4h,tlc检测反应完后,乙酸乙酯萃取,有机相用饱和氯化钠溶液洗涤,干燥,旋干,最后柱层析分离得产物1,产率65.0%,纯度95.6%;第二步:向烧瓶中加入中间体i和氢氧化钠,加入水搅拌,50℃反应10分钟,将第一步的产物与甲苯置于恒压滴定漏斗中,滴加到上述溶液中,滴加完保温50℃反应2小时,反应液用乙酸乙酯萃取,有机相用饱和氯化钠溶液洗涤,再干燥,用旋转蒸发仪蒸干,最后柱层析分离得产物2,产率77.8%,纯度98.9%。该技术方案的操作较多,并且整体产率仅有50%,使用柱层析不适合放大生产。具体合成路线如下:中国专利cn110028462a公开的氟雷拉纳合成方法分三步进行,第一步:向烧瓶中加入dmf、中间体i和ncs,搅拌,在40℃反应2小时。降温至室温,加水析出固体,继续降温至5℃,过滤,干燥得产物1,收率92.0%。第二步:向烧瓶中加入dmf,产物1和中间体ii,搅拌溶清后加入et3n室温反应10小时。向反应液中加入水,再用乙酸乙酯萃取体系三次,有机相合并,用无水硫酸钠干燥,然后用旋转蒸发仪蒸干得粗品,粗品再用乙酸乙酯和石油醚重结晶得产物2,纯度99.5%,收率85.4%。第三步:产物2与中间体iii进行酰胺化反应得产物3,即氟雷拉纳。具体合成路线如下:该技术方案此合成路线步骤较多,操作繁杂,有的中间体可以不用分离再进行下一步,放大生产会增加成本。因此,有必要研究一种操作简单、适合大生产的氟雷拉纳工艺合成路线,来满足市场需求。技术实现要素:为了克服上述现有技术的不足,本发明提供一种异噁唑啉类驱虫药的制备方法,该方法使用一种溶剂一锅法合成氟雷拉纳,减少了合成过程的工艺操作,缩短了处理时间和反应时间,同时提高了产品收率,适合大生产。为了实现上述目的,本发明采用如下技术方案:一种异噁唑啉类驱虫药的制备方法,其特征在于该异噁唑啉类驱虫药为氟雷拉纳,其结构式如化合物1所示,包括如下制备步骤:将中间体i在同一反应瓶内依次进行酰胺化缩合反应、取代反应、关环反应,反应完成后倒入水中,搅拌过滤,得到的固体经重结晶得到化合物1;其合成路线为:酰胺化缩合反应:将中间体i、缩合剂、中间体ii、有机碱加入反应容器中,在溶剂中进行反应,反应过程中用tlc检测反应程度,至原料中间体i反应完全;取代反应:向上述混合溶液中加入ncs进行反应,反应过程中用tlc检测反应程度,至产物1反应完全;关环反应:向取代反应后的混合溶液中加入中间体iii进行反应,反应过程中用tlc检测反应程度,至产物2反应完全。以中间体i为1.0当量,各反应物的摩尔比例分别为:ncs1.1~1.5,中间体ii1.1~1.5,有机碱2.0~3.5,缩合剂1.1~2.5,中间体iii1.1~1.5。所述取代反应中,将ncs溶解在溶剂中缓慢加入酰胺化缩合反应得到的混合溶液中。tlc检测使用的展开剂及体积比为pe:ea=1:1。各步骤的反应温度分别为20~30℃。酰胺化缩合反应中,所述缩合剂为dcc、bop-cl、pybop、cdi、dic、hobt与edc盐酸盐、hatu、hbtu、tbtu中的一种。酰胺化缩合反应中,所述有机碱为et3n、吡啶、dmap、diea中的一种。反应过程使用的溶剂为dmf、乙腈、二氯甲烷、dmso、nmp、dma中的一种。重结晶使用的溶剂为乙腈、甲醇、乙醇、异丙醇、四氢呋喃、二氯甲烷、正己烷、正庚烷、水、丙酮、甲苯、石油醚、乙酸乙酯中的至少一种。本发明的有益效果是:反应过程涉及的各步骤均使用同一种溶剂,并在在同一个反应瓶中进行,即使用一锅法合成氟雷拉纳,反应过程不需要分离中间产物,极大地减少了操作及处理步骤,同时整体反应过程耗时短,节省溶剂和时间,反应效率大大提高;此外,所得产物的摩尔收率高达80%,检测纯度在98.0%以上。本发明的合成方法得到的产物收率提高,且节约原料、节省时间。此方法合成路线简单,原料便宜,合成成本低,同时因为减少后处理操作而提高了收率,适合放大生产,可以满足市场的大量需求。附图说明图1为本发明实施例1所得产物的hplc谱图;图2为本发明实施例1所得产物的1hnmr谱图。具体实施方式为了使本发明的目的、技术方案和优点更加清楚,下面将结合附图对本发明作进一步地详细描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其它实施例,都属于本发明保护的范围。实施例1:第一步:酰胺化缩合反应向500ml四口瓶中加入200mldmf,搅拌,再加入20g(0.11mol)中间体i,控温20~30℃,向反应液中加入18.0g(0.13mol)hobt,25.6g(0.13mol)edc盐酸盐和30g(0.16mol)中间体ii,再加入25.9g(0.26mol)et3n,保温反应2小时,使用pe:ea=1:1(体积比)的展开剂进行tlc检测,至中间体i反应完全。第二步:取代反应再向上述反应液中分批缓慢加入19.4g(0.15mol)ncs,加完后在20~30℃保温反应2小时,tlc(pe:ea=1:1)检测产物1反应完全。第三步:关环反应向第二步得到的反应液中加入32.2g(0.13mol)中间体iii,保温反应3小时,tlc检测(pe:ea=1:1)产物2反应完全。将反应液缓慢加入到2l纯化水中,边加边搅拌,加完后搅拌1小时,过滤。固体用石油醚与乙酸乙酯重结晶,过滤,晾干得产品47.8g,摩尔收率83.0%。为了说明得到的产物的结构,将得到的产物进行1hnmr测试,使用的仪器型为brukeravanceiii400mhz,样品使用cdcl3溶解,得到的核磁谱图如图1所示,图1中各峰的位置强弱为:δ7.36-7.53(m,6h),7.07(t,1h),6.76(t,1h),4.21(t,2h),4.06(t,1h),3.94(m,2h),3.72(d,1h),2.46(s,1h)。对各峰进行分析,其中,7.36-7.53ppm为两个苯环上所有的氢,7.07ppm的峰是与三氟乙基相连的n上的氢,6.76ppm的峰是苯甲酰基上的n上的氢,4.21ppm的峰是三氟乙基上亚甲基的氢,3.94ppm的峰是与苯甲酰胺相连的亚甲基的氢,3.72ppm与4.06ppm的峰是异恶唑上亚甲基的两个氢,由于与异恶唑亚甲基相邻的炭具有手性,所以异恶唑亚甲基的两个氢ppm不同,2.45ppm的峰是苯环上的甲基上的氢。根据以上分析,核磁氢谱与氟雷拉纳结构吻合,确认所得产物为氟雷拉纳。为了对所得产物的纯度进行分析,将所得产物进行hplc测试,使用的色谱仪和色谱住相关信息见表1。表1仪器型号与规格厂家高效液相色谱仪lc-20at,spd-20a,sil-20ashimadzu电子天平es225sm-drprecisa色谱柱ultimatexbc18(4.6mm×250mm,5μm)月旭科技(上海)股份有限公司hplc测试的检测条件为:用十八烷基硅烷键合硅胶为填充剂(月旭ultimatexbc18柱,4.6mm×250mm,5μm或效能相当的色谱柱);以0.2%乙酸溶液:乙腈(35:65)为流动相;流速为每分钟1.0ml;检测波长为254nm;进样量为20μl。试样处理方式:称取10mg固体,用1ml乙腈溶解,进样20μl。测得的液相色谱图如图2所示,从图2可以得出,所得产物的纯度为99.8%。实施例2:向500ml四口瓶中加入200mldmso,搅拌,再加入20g(0.11mol)中间体i,控温20~30℃向反应液中加入18.0g(0.13mol)hobt,25.6g(0.13mol)edc盐酸盐和30g(0.16mol)中间体ii,再加入25.9g(0.26mol)et3n,保温反应2小时,tlc检测(pe:ea=1:1)至中间体i反应完全。再向反应液中分批缓慢加入19.4g(0.15mol)ncs,加完20~30℃保温反应2小时,tlc检测(pe:ea=1:1)。向反应液中加入32.3g(0.13mol)中间体iii,保温反应3小时,tlc检测(pe:ea=1:1)。将反应液缓慢加入到2.8l纯化水中,边加边搅拌,加完后搅拌1小时,过滤。固体用石油醚与乙酸乙酯重结晶,过滤,晾干得44g,摩尔收率79.1%,hplc检测纯度98.7%。实施例3:向500ml四口瓶中加入200mldmf,搅拌,再加入20g(0.11mol)中间体i,控温20~30℃向反应液中加入37.8g(0.28mol)hobt,53.48g(0.28mol)edc盐酸盐和32.6g(0.17mol)中间体ii,再加入39.59g(0.39mol)et3n,保温反应2小时,tlc检测(pe:ea=1:1)至中间体i反应完全。再向反应液中分批缓慢加入22.4g(0.17mol)ncs,加完20~30℃保温反应2小时,tlc检测(pe:ea=1:1)。向反应液中加入40.5g(0.17mol)中间体iii,保温反应3小时,tlc检测(pe:ea=1:1)。将反应液缓慢加入到2.8l纯化水中,边加边搅拌,加完后搅拌1小时,过滤。固体用甲苯与乙酸乙酯重结晶,过滤,晾干得42.6g,摩尔收率76.5%,hplc检测纯度98.5%。实施例4:向500ml四口瓶中加入200mldmf,搅拌,再加入20g(0.11mol)中间体i,控温20~30℃向反应液中加入16.6g(0.12mol)hobt,23.5g(0.12mol)edc盐酸盐和23.6g(0.12mol)中间体ii,再加入22.6g(0.22mol)et3n,保温反应2小时,tlc检测(pe:ea=1:1)至中间体i反应完全。再向反应液中分批缓慢加入16.4g(0.12mol)ncs,加完在20~30℃保温反应2小时,tlc检测(pe:ea=1:1)至反应完全。向反应液中加入29.7g(0.12mol)中间体iii,保温反应3小时,tlc检测(pe:ea=1:1)至反应完全。将反应液缓慢加入到2l纯化水中,边加边搅拌,加完后搅拌1小时,过滤。固体用石油醚与乙酸乙酯重结晶,过滤,晾干得43.0g,摩尔收率77..4%,hplc检测纯度99.0%。实施例5:向500ml四口瓶中加入200mlnmp,搅拌,再加入20g(0.11mol)中间体i,控温20~30℃向反应液中加入16.6g(0.12mol)hobt,23.5g(0.12mol)edc盐酸盐和23.6g(0.12mol)中间体ii,再加入17.7g(0.22mol)吡啶,保温反应2小时,tlc检测(pe:ea=1:1)至中间体i反应完全。再向反应液中分批缓慢加入16.4g(0.12mol)ncs,加完20~30℃保温反应2小时,tlc检测(pe:ea=1:1)至反应完全。向反应液中加入29.7g(0.123mol)中间体iii,保温反应3小时,tlc检测(pe:ea=1:1)至反应完全。将反应液缓慢加入到2l纯化水中,边加边搅拌,加完后搅拌1小时,过滤。固体用石油醚与乙酸乙酯重结晶,过滤,晾干得40.0g,摩尔收率63.3%,hplc检测纯度98.0%。实施例6:向500ml四口瓶中加入200mldmf,搅拌,再加入20g(0.11mol)中间体i,控温20~30℃向反应液中加入19.9g(0.12mol)cdi,室温反应1小时,加入23.6g(0.12mol)中间体ii,保温反应2小时,tlc检测(pe:ea=1:1)至中间体i反应完全。再向反应液中分批缓慢加入16.4g(0.123mol)ncs,加完20~30℃保温反应2小时,tlc检测(pe:ea=1:1)至反应完全。向反应液中加入29.7g(0.12mol)中间体iii,保温反应3小时,tlc检测(pe:ea=1:1)至反应完全。将反应液缓慢加入到2l纯化水中,边加边搅拌,加完后搅拌1小时,过滤。固体用甲苯与乙酸乙酯重结晶,过滤,晾干得35.8g,摩尔收率57.4%,hplc检测纯度96.0%。以上对本发明的实例进行了详细说明,但所述内容仅为本发明的较佳实施例,不能被认为用于限定本发明的实施范围。凡依本发明申请范围所作的均等变化与改进等,均应仍归属于本发明的专利涵盖范围之内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1