喹唑啉类化合物及其药物组合物的制作方法
1.本技术涉及生物医药领域,具体的涉及一种喹唑啉类化合物及其药物组合物。
背景技术:
2.hpk1是英文hematopoietic progenitor kinase 1的缩写,即造血祖细胞激酶1,隶属于丝氨酸/苏氨酸激酶家族,又名map4k1,即mitogen-activated protein kinase 1。hpk1主要表达于淋巴器官,具体包括骨髓、胎儿肝脏、淋巴结、胎盘、脾脏与胸腺(adv immunol,2016,129:277-314),在人体主要组织中均无分布,因此,预计选择性作用于该靶点的激酶抑制剂安全性相对更高。
技术实现要素:
3.本技术的一个目的是提供一种喹唑啉类化合物及其药物组合物,本技术的化合物可高效抑制hpk1激酶的活性,具备极高的成药性。
4.一方面,本技术提供了一种式i的化合物:
[0005][0006]
或其药学上可接受的盐、溶剂化物、立体异构体、前药、代谢物或衍生物,其中,ra,rb,rc,rd之一为其余各自独立地选自h、羟基、卤素、氰基、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代的烷基、c
3-8
取代的环烷基和c
1-6
烷氧基;
[0007]
re选自h、羟基、卤素、氰基、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代的烷基、c
3-8
取代的环烷基和c
1-6
烷氧基;
[0008]
x选自—o—、—nh—、—s—、—so—、—so2—、羰基、羰基氨基和氨基羰基;
[0009]
r1为取代的环烷基、取代的杂环基或者m为0、1、2、3、4或5,r
14
为c
1-6
烷基、c
3-8
环烷基、c
1-6
取代的烷基或c
3-8
取代的环烷基;
[0010]
a环为取代的苯环或取代的5-6元芳香杂环,其中,所述取代的5-6元芳香杂环具有1-4个选自o、s和n的杂原子;
[0011]
r2选自以下取代基:
[0012][0013]
其中y为c或者n,r7为h、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代的烷基或者c
3-8
取代的环烷基,r8为h、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代的烷基或者c
3-8
取代的环烷基,r9和r
10
独立地为h、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代的烷基或者c
3-8
取代的环烷基,r
11
为h、氨基、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代的烷基或者c
3-8
取代的环烷基,r
12
为4-6元杂环基或者4-6取代的杂环基。
[0014]
在一些实施方式中,所述r1为时,m取值为1或者2。
[0015]
在一些实施方式中,所述r
14
为c
1-6
烷基。
[0016]
在一些实施方式中,所述r1为取代的5-8元环烷基或取代的5-8元杂环基,其中所述取代的5-8元杂环基具有1-4个选自o、s和n的杂原子。
[0017]
在一些实施方式中,所述r1为取代的5-6元环烷基或取代的5-6元杂环基,其中所述取代的5-6元杂环基具有1-4个选自o、s和n的杂原子。
[0018]
在一些实施方式中,当r1为取代的5-8元环烷基时,所述r1为其中n为1、2、3或4。
[0019]
在一些实施方式中,所述r3为—or4或者—nr5r6,其中r4为h、烷基、取代的烷基、环烷基或者取代的环烷基,r5和r6独立地为h、烷基、取代的烷基、环烷基或者取代的环烷基,其中r5和r6不同时为环烷基或者取代的环烷基。
[0020]
在一些实施方式中,所述r4为h、c
1-6
烷基、c
1-6
取代烷基、c
3-8
环烷基或者c
3-8
取代的环烷基。
[0021]
在一些实施方式中,所述r5和r6独立地为h、c
1-6
烷基、c
1-6
取代烷基、c
3-8
环烷基或者c
3-8
取代的环烷基,其中r5和r6不同时为c
3-8
环烷基或者c
3-8
取代的环烷基。
[0022]
在一些实施方式中,所述r
13
为h、c
1-6
烷基、c
1-6
取代烷基、c
3-8
环烷基或者c
3-8
取代的环烷基。
[0023]
在一些实施方式中,r1选自以下取代基:
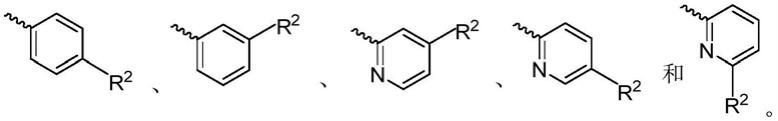
[0024][0025]
在一些实施方式中,a环选自以下取代芳香环:
[0026][0027]
在一些实施方式中,所述a环为取代的苯环、取代的吡啶环或取代的吡唑环。
[0028]
在一些实施方式中,当a环为取代的苯环或者取代的吡啶环时,r2取代位置位于x的对位或者间位。
[0029]
在一些实施方式中,r2选自以下取代基:
[0030][0031]
在一些实施方式中,r
12
选自以下取代基:
[0032][0033]
在一些实施方式中,所述的式i的化合物如式i-1所示,
[0034][0035]
或其药学上可接受的盐、溶剂化物、立体异构体、前药、代谢物或衍生物,其中,rb,rc,rd,re各自独立地选自h、羟基、卤素、氰基、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代的烷基、c
3-8
取代的环烷基、c
1-6
烷氧基;
[0036]
x选自—o—、—nh—、—s—、—so—、—so2—、羰基、羰基氨基和氨基羰基;
[0037]
r1为取代的环烷基、取代的杂环基或者m为0、1、2、3、4或5,r
14
为c
1-6
烷基、c
3-8
环烷基、c
1-6
取代的烷基或c
3-8
取代的环烷基;
[0038]
a环为取代的苯环或取代的5-6元芳香杂环,其中,所述取代的5-6元芳香杂环具有1-4个选自o、s和n的杂原子;
[0039]
r2选自以下取代基:
[0040][0041]
其中y为c或者n,r7为h、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代烷基或者c
3-8
取代的环烷基,r8为h、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代烷基或者c
3-8
取代的环烷基,r9和r
10
独立地为h、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代烷基或者c
3-8
取代的环烷基,r
11
为h、氨基、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代烷基或者c
3-8
取代的环烷基,r
12
为4-6元取代杂环基或者4-6元未取代杂环基。
[0042]
在一些实施方式中,所述的式i的化合物如式i-1-1或i-1-2所示,
[0043][0044]
或其药学上可接受的盐、溶剂化物、立体异构体、前药、代谢物或衍生物,其中,rb,rc,rd,re各自独立地选自h、羟基、卤素、氰基、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代的烷基、c
3-8
取代的环烷基和c
1-6
烷氧基;
[0045]
x选自—o—、—nh—、—s—、—so—、—so2—、羰基、羰基氨基和氨基羰基;
[0046]
n为1、2、3或4;
[0047]
m为0、1、2、3、4或5;
[0048]r14
为c
1-6
烷基、c
3-8
环烷基、c
1-6
取代的烷基或c
3-8
取代的环烷基;
[0049]
a环为取代的苯环或取代的5-6元芳香杂环,其中,所述取代的5-6元芳香杂环具有1-4个选自o、s和n的杂原子;
[0050]
r2选自以下取代基:
[0051][0052]
其中y为c或者n,r7为h、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代烷基或者c
3-8
取代的环烷基,r8为h、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代烷基或者c
3-8
取代的环烷基,r9和r
10
独立地为h、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代烷基或者c
3-8
取代的环烷基,r
11
为h、氨基、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代烷基或者c
3-8
取代的环烷基,r
12
为4-6元取代杂环基或者4-6元未取代杂环基。
[0053]
在一些实施方式中,所述式i的化合物选自以下化合物:
[0054]
[0055]
[0056]
[0057][0058][0059]
在一些实施方式中,所述式i的化合物选自以下化合物:
[0060]
8-(((1s,4s)-4-氨基环己基)氧代)-n-(1-(1-甲基哌啶-4-基)-1h-吡唑-4-基)喹唑啉-2-胺;
[0061]
4-((2-((4-((2-(二甲氨基)乙基)(甲基)氨基)苯基)氨基)喹唑啉-8-基)氧代)环己烷-1-醇;
[0062]
4-((2-((1-(1-甲基哌啶-4-基)-1h-吡唑-4-基)氨基)喹唑啉-8-基)氧代)环己烷-1-醇;
[0063]
4-((2-((4-(4-甲基哌嗪-1-基)苯基)氨基)喹唑啉-8-基)氧代)环己烷-1-醇;
[0064]
8-(2-甲氧基乙氧基)-n-(4-(4-甲基哌嗪-1-基)苯基)喹唑啉-2-胺;
[0065]
3-((2-((4-(4-甲基哌嗪-1-基)苯基)氨基)喹唑啉-8-基)氧代)环戊烷-1-醇;
[0066]
4-((2-((5-(4-甲基哌嗪-1-基)吡啶-2-基)氨基)喹唑啉-8-基)氧代)环己烷-1-醇;
[0067]
4-((8-((4-羟基环己基)氧代)喹唑啉-2-基)氨基)-n-(1-甲基哌啶-4-基)苯酰胺;
[0068]
4-((8-((3-羟基环戊基)氧代)喹唑啉-2-基)氨基)苯磺酰胺;和
[0069]
3-((8-((4-羟基环己基)氧代)喹唑啉-2-基)氨基)苯磺酰胺。
[0070]
另一方面,本技术提供了一种药物组合物,其包含所述的式i的化合物,或其药学上可接受的盐、溶剂化物、立体异构体、前药、代谢物或衍生物;和药学上可接受的载体。
[0071]
本领域技术人员能够从下文的详细描述中容易地洞察到本技术的其它方面和优势。下文的详细描述中仅显示和描述了本技术的示例性实施方式。如本领域技术人员将认识到的,本技术的内容使得本领域技术人员能够对所公开的具体实施方式进行改动而不脱离本技术所涉及发明的精神和范围。相应地,本技术的附图和说明书中的描述仅仅是示例性的,而非为限制性的。
附图说明
[0072]
本技术所涉及的发明的具体特征如所附权利要求书所显示。通过参考下文中详细描述的示例性实施方式和附图能够更好地理解本技术所涉及发明的特点和优势。对附图简要说明书以下:
[0073]
图1显示的是本技术所述化合物dd02001h的合成路线示意图;
[0074]
图2显示的是本技术所述化合物dd02013h的合成路线示意图;
[0075]
图3显示的是本技术所述化合物dd02014h的合成路线示意图;
[0076]
图4显示的是本技术所述化合物dd02006h的合成路线示意图;
[0077]
图5显示的是本技术所述化合物dd02008h的合成路线示意图;
[0078]
图6显示的是本技术所述化合物dd02015h的合成路线示意图;
[0079]
图7显示的是本技术所述化合物dd02021h的合成路线示意图;
[0080]
图8显示的是本技术所述化合物dd02018h的合成路线示意图;
[0081]
图9显示的是本技术所述化合物dd02002h的合成路线示意图;
[0082]
图10显示的是本技术所述化合物dd02019h的合成路线示意图。
具体实施方式
[0083]
以下由特定的具体实施例说明本技术的实施方式,熟悉此技术的人士可由本说明书所公开的内容容易地了解本技术的其他优点及效果。
[0084]
术语定义
[0085]
术语“取代基”或“取代基团”是指替代分子上氢原子的原子或原子团。术语“取代的”是指特定的分子带有一个或多个取代基。
[0086]
术语“烷基”是指支链和直链的饱和脂族烃基,其含有例如1至12个碳原子、1至6个碳原子或1至4个碳原子。烷基基团的实例包括但不限于甲基(me)、乙基(et)、丙基(例如正丙基和异丙基)、丁基(例如正丁基、异丁基、仲丁基和叔丁基)以及戊基(例如正戊基、异戊基、新戊基)、正己基、2-甲基戊基、2-乙基丁基、3-甲基戊基和4-甲基戊基。当数字以下标的形式出现在符号“c”后时,所述下标更具体地定义特定基团可含有的碳原子的数目。例如,“c
1-6
烷基”表示具有一至六个碳原子的直链和支链烷基基团。c
1-6
烷基的实例包括甲基、乙基、丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基和戊基。
[0087]
本技术使用的术语“环烷基”是指通过由饱和环碳原子移去一个氢原子由非芳族单环烃分子衍生得到的基团。环烷基基团的代表性实例包括但不限于环丙基、环戊基和环己基。当数字以下标形式出现在符号“c”后时,所述下标更具体地定义特定的环烷基基团可含有的碳原子的数目。例如,“c
3-6
环烷基”表示具有三至六个碳原子的环烷基基团。单环的环烷基的实例是环丙基、环丁基、环戊基、环己基或环庚基。
[0088]
术语“杂原子”是指氧(o)、硫(s)和氮(n)。
[0089]
术语“卤代”和“卤素”是指f、cl、br和i。
[0090]
术语“氰基”是指基团-cn。
[0091]
术语“氨基”是指基团-nh2。
[0092]
术语“杂环烷基”或“杂环基”是指非芳族3-8元单环、7-12元双环或10-14元三环环系,其包含1-4个杂原子(若为单环的)、1-6个杂原子(若为双环的)或1-9个杂原子(若为三环的),所述杂原子选自o、s或n,其中该非芳族环体系为完全饱和的。杂环烷基基团可任选地经一个或多个取代基取代。在一个实施方案中,杂环烷基基团各个环的0个、1个、2个、3个或4个原子可被取代基取代。
[0093]
术语“芳香环”或“芳基”是指烃的单环、双环或三环芳族环体系。芳基基团可任选地经一个或多个取代基取代。在一个实施方案中,芳基基团各个环的0个、1个、2个、3个、4个、5个或6个原子可被取代基取代。芳基基团的实例包括苯基、萘基、蒽基、芴基、茚基、薁基,等等。
[0094]
术语“芳香杂环”或“杂芳基”是指取代的和未取代的芳族5或6元单环基团和9或10元双环基团,所述基团在至少一个环中具有至少一个杂原子(o、s或n),所述含有杂原子的环优选具有1、2或3个独立地选自o、s和/或n的杂原子。含有杂原子的杂芳基基团中的每个环可以含有1或2个氧或硫原子和/或1-4个氮原子,条件是在每个环中杂原子的总数为4或更少并且每个环具有至少一个碳原子。杂芳基基团可以连接于任何环的任何可利用的氮或碳原子上。杂芳基环系统可以是未取代的或可以含有一个或多个取代基。
[0095]
术语“烷氧基”是指-o-烷基基团。烷氧基基团可任选地经一个或多个取代基取代。
[0096]
术语“异构体”或“立体异构体”是指具有相同化学组成但原子或基团在空间中的排列不同的化合物。
[0097]
术语“药学上可接受的”是指这样的化合物、物质、组合物和/或剂型,其在合理的医药判断范围内适用于与人类和动物的组织接触而不引起过度的毒性、刺激性、过敏反应
或其它问题或并发症,这与合理的益处/风险比例相称。
[0098]
式i的化合物可以形成盐,这些盐也在本技术的范围内。除非另有指示,否则提及本技术化合物应理解为包括提及其一种或多种盐。术语“盐”表示与无机和/或有机酸以及碱形成的酸性和/或碱性盐。此外,术语“盐”可包括两性离子(内盐),例如,当式i的化合物含有碱性部分(例如胺或吡啶或咪唑环)以及酸性部分(例如羧酸)时。优选药学上可接受的(即,无毒、生理上可接受的)盐,例如可接受的金属盐和胺盐,其中阳离子对该盐的毒性或生物活性无显著贡献。然而,其它盐也可用于例如制备期间可所采用的分离或纯化步骤中,且因而涵盖于本技术范围内。式i化合物的盐可通过,例如,将式i的化合物与一定量(如一当量)的酸或碱在诸如该盐在其中沉淀的介质或在水性介质中反应,然后冻干来形成。
[0099]
应进一步理解,式i的化合物的溶剂化物(例如,水合物)也在本技术的范围内。术语“溶剂化物”是指式i的化合物与一种或多种溶剂分子(不论有机或无机)的物理性缔合。此物理性缔合包括氢键合。在某些情况下,溶剂化物将是能够分离的,例如当一个或多个溶剂分子掺入结晶固体的晶格中时。示例性的溶剂化物包括水合物、乙醇化物、甲醇化物、异丙醇化物、乙腈溶剂化物和乙酸乙酯溶剂化物。溶剂化方法在本领域中是已知的。
[0100]
术语“代谢物”是指通过在体内代谢特定化合物或其盐而产生的产物。可使用本领域已知的常规技术鉴定化合物的代谢物,并使用如本技术所述的那些技术确定它们的活性。此产物可由例如所述给药的化合物的氧化、羟基化、还原、水解、酰胺化、脱酰胺化、酯化、脱酯化、酶切等产生。因此,本技术包括本技术化合物的代谢物,包括通过以下方法产生的化合物,该方法包括使本技术化合物与哺乳动物接触足以产生其代谢产物的时间段。
[0101]
术语“前药”或“前体药物”通常是指药物前体的化合物,其在给予受试者时,通过代谢或化学过程经历化学转化,得到式i化合物或其盐。关于前药的内容是本领域所熟知的(参见,例如berge et al.(1977)"pharmaceutical salts",j.pharm.sci.66:1-19)。
[0102]
前药部分的实例包括经取代和未经取代的、支链的或无支链的低级烷基酯部分(例如,丙酸酯)、低级烯基酯、二-低级烷基-氨基低级烷基酯(例如,二甲基氨基乙基酯)、酰氨基低级烷基酯(例如,乙酰氧基甲基酯)、酰氧基低级烷基酯(例如,新戊酰氧基甲基酯)、芳基酯(苯基酯)、芳基-低级烷基酯(例如,苄基酯)、经取代的(例如,经甲基、卤素或甲氧基取代基取代的)芳基和芳基-低级烷基酯、酰胺、低级烷基酰胺、二-低级烷基酰胺和羟基酰胺。还包括通过体内其它机制转化为活性形式的前药。
[0103]
式i的化合物可以形成酯,这些酯也在本技术的范围内。术语“酯”是指其在体内水解并且包括在人类体内容易分解以留下母体化合物或其盐的那些酯。合适的酯基包括,举例来说,从药学上可接受的脂肪族羧酸衍生得到的那些酯基。具体酯的代表性的例子包括但不限于,甲酸酯、乙酸酯、丙酸酯、丁酸酯、丙烯酸酯和丁二酸乙酯。
[0104]
此外,式i的化合物在它们的制备后可以进行分离和纯化,得到含有按重量计的量等于或大于99%的式i的化合物(“基本纯的”)的组合物,然后将其如本技术所述使用或配制。此种“基本纯的”式i的化合物在本技术中也被认为是本技术的一部分。
[0105]
本技术化合物意在包括本技术化合物中出现的原子的所有同位素。同位素包括具有相同原子数而不同质量数的原子。作为一般实例而非限制,氢的同位素包括氘(d)和氚(t)。碳的同位素包括
13
c和
14
c。同位素标记的本技术化合物通常可通过本领域技术人员已知的常规技术或通过类似于本技术所述的方法使用适当的同位素标记的试剂替代在其它
情况下所采用的未经标记的试剂来制备。
[0106]
术语“衍生物”指母体化合物分子中的原子或原子团被其他原子或原子团取代所形成的化合物,称为该母体化合物的衍生物。如卤代烃、醇、醛、羧酸可看成是烃的衍生物,因为它们是烃的氢原子被取代为卤素、羟基、氧等的产物。又如酰卤、酸酐、酯是羧酸衍生物,因为它们是羧酸中的羟基被卤素和一些有机基团取代的产物。例如,以甲烷(ch4)为母体,则甲醇(ch3oh)、甲酸(hcooh)、一氯甲烷(ch3cl)等均为甲烷的衍生物。
[0107]
术语“载体”包括药学上可接受的载体、赋形剂或稳定剂,它们对所用剂量和浓度的细胞或哺乳动物是无毒的。所述生理学上可接受的载体通常为ph缓冲水溶液。生理学上可接受的载体的非限制性实例包括缓冲剂,例如磷酸盐、柠檬酸盐和其它有机酸;抗氧化剂,包括抗坏血酸;低分子量(小于约10个残基)多肽;蛋白质,例如血清白蛋白、明胶或免疫球蛋白;亲水性聚合物,例如聚乙烯基吡咯烷酮;氨基酸,例如甘氨酸、谷氨酰胺、天冬酰胺、精氨酸或赖氨酸;单糖、二糖和其它碳水化合物(包括葡萄糖、甘露糖或糊精);螯合剂如edta;糖醇如甘露醇或山梨糖醇;成盐的抗衡离子如钠;以及/或非离子表面活性剂,例如tween
tm
、聚乙二醇(peg)和pluronics
tm
。在某些实施方案中,所述药学上可接受的载体为非天然存在的药学上可接受的载体。
[0108]
术语“抑制”意指,与不存在该抑制剂时所述酶的活性相比,降低该目标酶的活性。在一些实施方案中,术语“抑制”意指hpk1活性降低至少约5%、至少约10%、至少约20%、至少约25%、至少约50%、至少约60%、至少约70%、至少约80%、至少约90%或至少约95%。在其它实施方案中,抑制意指hpk1活性降低约5%至约25%、约25%至约50%、约50%至约75%或约75%至100%。在一些实施方案中,抑制意指hpk1活性降低约95%至100%,例如活性降低95%、96%、97%、98%、99%或100%。可使用各种技术测量这种降低,这些技术为本领域技术人员可掌握的,其包括体外激酶测定。
[0109]“hpk1拮抗剂”或“hpk1抑制剂”为一种分子,该分子降低、抑制或以其它方式减少hpk1的一种或多种生物活性(例如,丝氨酸/苏氨酸激酶活性、在tcr激活后募集到tcr复合物、与蛋白质结合配偶体(例如slp76)相互作用)。使用所述hpk1拮抗剂的拮抗作用不一定表明完全消除hpk1活性。相反,该活性可减少统计学上显著的量,包括例如,与适当的对照相比,减少hpk1活性的至少约5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、95%或100%。
[0110]
发明详述
[0111]
喹唑啉类化合物
[0112]
一方面,本技术提供了式i的化合物:
[0113][0114]
或其药学上可接受的盐、溶剂化物、立体异构体、前药、代谢物或衍生物,其中,ra,
rb,rc,rd之一可以为其余可以各自独立地选自h、羟基、卤素、氰基、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代的烷基、c
3-8
取代的环烷基和c
1-6
烷氧基;
[0115]
re可以选自h、羟基、卤素、氰基、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代的烷基、c
3-8
取代的环烷基和c
1-6
烷氧基;
[0116]
x可以选自—o—、—nh—、—s—、—so—、—so2—、羰基、羰基氨基和氨基羰基;
[0117]
r1可以为取代的环烷基、取代的杂环基或者m可以为0、1、2、3、4或5,r
14
可以为c
1-6
烷基、c
3-8
环烷基、c
1-6
取代的烷基或c
3-8
取代的环烷基;
[0118]
a环可以为取代的苯环或取代的5-6元芳香杂环,其中,所述取代的5-6元芳香杂环具有1-4个选自o、s和n的杂原子;
[0119]
r2可以选自以下取代基:
[0120][0121]
其中y可以为c或者n,r7可以为h、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代的烷基或者c
3-8
取代的环烷基,r8可以为h、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代的烷基或者c
3-8
取代的环烷基,r9和r
10
可以独立地为h、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代的烷基或者c
3-8
取代的环烷基,r
11
可以为h、氨基、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代的烷基或者c
3-8
取代的环烷基,r
12
可以为4-6元杂环基或者4-6取代的杂环基。
[0122]
在本技术中,ra,rb,rc,rd之一可以为其余可以各自独立地选自h、羟基、卤素、氰基、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代的烷基、c
3-8
取代的环烷基和c
1-6
烷氧基表示这样一类结构:当ra,rb,rc,rd的其中一个为时,其余三个可以各自独立地选自h、羟基、卤素、氰基、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代的烷基、c
3-8
取代的环烷基和c
1-6
烷氧基;例如,当ra为时,rb,rc,rd可以各自独立地选自h、羟基、卤素、氰基、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代的烷基、c
3-8
取代的环烷基和c
1-6
烷氧基。
[0123]
在一些实施方式中,所述r1可以为时,m取值为1或者2。
[0124]
在一些实施方式中,所述r
14
可以为c
1-6
烷基。
[0125]
在一些实施方式中,所述r1可以为取代的5-8元环烷基或取代的5-8元杂环基,其中所述取代的5-8元杂环基具有1-4个选自o、s和n的杂原子。
[0126]
在一些实施方式中,所述r1可以为取代的5-6元环烷基或取代的5-6元杂环基,其中所述取代的5-6元杂环基具有1-4个选自o、s和n的杂原子。
[0127]
在一些实施方式中,当r1为取代的5-8元环烷基时,所述r1可以为其中n可以为1、2、3或4。
[0128]
在一些实施方式中,所述r3可以为—or4或者—nr5r6,其中r4可以为h、烷基、取代的烷基、环烷基或者取代的环烷基,r5和r6可以独立地为h、烷基、取代的烷基、环烷基或者取代的环烷基,其中r5和r6不同时为环烷基或者取代的环烷基。
[0129]
在一些实施方式中,所述r4可以为h、c
1-6
烷基、c
1-6
取代烷基、c
3-8
环烷基或者c
3-8
取代的环烷基。
[0130]
例如,当r4为h时,r3为—oh,r1可以为其中n可以为1、2、3或4。
[0131]
在一些实施方式中,所述r5和r6可以独立地为h、c
1-6
烷基、c
1-6
取代烷基、c
3-8
环烷基或者c
3-8
取代的环烷基,其中r5和r6不同时为c
3-8
环烷基或者c
3-8
取代的环烷基。
[0132]
例如,当r5和r6均为h时,r3为—nh2,r1可以为其中n为1、2、3或4。
[0133]
在一些实施方式中,所述r
13
可以为h、c
1-6
烷基、c
1-6
取代烷基、c
3-8
环烷基或者c
3-8
取代的环烷基。
[0134]
在一些实施方式中,当r4为时,r3可以为
[0135]
例如,当r
13
为甲基时,r3为r1为其中n可以为1、2、3或4。
[0136]
在一些实施方式中,当r5为h,r6为时,r3可以为
[0137]
例如,当r
13
为甲基时,r3为r1为其中n可以为1、2、3或4。
[0138]
在一些实施方式中,r1可以选自以下取代基:
[0139][0140]
在一些实施方式中,a环可以选自以下取代芳香环:
[0141][0142]
在一些实施方式中,所述a环可以为取代的苯环、取代的吡啶环或取代的吡唑环。
[0143]
在一些实施方式中,当a环为取代的苯环或者取代的吡啶环时,r2取代位置可以位于x的对位或者间位。
[0144]
例如,a环可以选自以下结构:
[0145][0146]
在一些实施方式中,r2可以选自以下取代基:
[0147][0148]
例如,a环可以选自以下结构:
[0149][0150]
在一些实施方式中,a环可以选自以下结构:
[0151][0152]
在一些实施方式中,r
12
可以选自以下取代基:
[0153][0154]
例如,a环可以选自以下结构:
[0155][0156]
在一些实施方式中,所述的式i的化合物如式i-1所示,
[0157][0158]
或其药学上可接受的盐、溶剂化物、立体异构体、前药、代谢物或衍生物,其中,rb,rc,rd,re可以各自独立地选自h、羟基、卤素、氰基、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代的烷基、c
3-8
取代的环烷基和c
1-6
烷氧基;
[0159]
x可以选自—o—、—nh—、—s—、—so—、—so2—、羰基、羰基氨基和氨基羰基;
[0160]
r1可以为取代的环烷基、取代的杂环基或者m可以为0、1、2、3、4或5,r
14
可以c
1-6
烷基、c
3-8
环烷基、c
1-6
取代的烷基或c
3-8
取代的环烷基;
[0161]
a环可以为取代的苯环或取代的5-6元芳香杂环,其中,所述取代的5-6元芳香杂环具有1-4个选自o、s和n的杂原子;
[0162]
r2可以选自以下取代基:
[0163][0164]
其中y可以为c或者n,r7为h、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代烷基或者c
3-8
取代的环烷基,r8可以为h、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代烷基或者c
3-8
取代的环烷基,r9和r
10
可以独立地为h、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代烷基或者c
3-8
取代的环烷基,r
11
可以为h、氨基、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代烷基或者c
3-8
取代的环烷基,r
12
可以为4-6元取代杂环基或者4-6元未取代杂环基。
[0165]
在一些实施方式中,所述的式i的化合物可以如式i-1-1或i-1-2所示,
[0166][0167]
或其药学上可接受的盐、溶剂化物、立体异构体、前药、代谢物或衍生物,其中,rb,rc,rd,re可以各自独立地选自h、羟基、卤素、氰基、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代的烷基、c
3-8
取代的环烷基和c
1-6
烷氧基;
[0168]
x可以选自—o—、—nh—、—s—、—so—、—so2—、羰基、羰基氨基和氨基羰基;
[0169]
n可以为1、2、3或4;
[0170]
m可以为0、1、2、3、4或5;
[0171]r14
可以为c
1-6
烷基、c
3-8
环烷基、c
1-6
取代的烷基或c
3-8
取代的环烷基;
[0172]
a环可以为取代的苯环或取代的5-6元芳香杂环,其中,所述取代的5-6元芳香杂环具有1-4个选自o、s和n的杂原子;
[0173]
r2可以选自以下取代基:
[0174][0175]
其中y可以为c或者n,r7可以为h、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代烷基或者c
3-8
取代的环烷基,r8可以为h、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代烷基或者c
3-8
取代的环烷基,r9和r
10
可以独立地为h、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代烷基或者c
3-8
取代的环烷基,r
11
可以为h、氨基、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代烷基或者c
3-8
取代的环烷基,r
12
可以为4-6元取代杂环基或者4-6元未取代杂环基。
[0176]
在一些实施方式中,所述r3可以为—or4或者—nr5r6,其中r4可以为h、烷基、取代的烷基、环烷基或者取代的环烷基,r5和r6可以独立地为h、烷基、取代的烷基、环烷基或者取代的环烷基,其中r5和r6不同时为环烷基或者取代的环烷基。
[0177]
在一些实施方式中,所述r4可以为h、c
1-6
烷基、c
1-6
取代烷基、c
3-8
环烷基或者c
3-8
取代的环烷基。
[0178]
例如,当r4为h时,r3为—oh,r1可以为其中n为1、2、3或4,所述的式i的化合物可以如式i-1-1a所示,
[0179][0180]
在一些实施方式中,所述r5和r6可以独立地为h、c
1-6
烷基、c
1-6
取代烷基、c
3-8
环烷基或者c
3-8
取代的环烷基,其中r5和r6不同时为c
3-8
环烷基或者c
3-8
取代的环烷基。
[0181]
例如,当r5和r6均为h时,r3为—nh2,r1可以为其中n为1、2、3或4,所述的式i的化合物可以如式i-1-1b所示,
[0182]
[0183]
在一些实施方式中,当r4为时,r3可以为所述的式i的化合物可以如式i-1-1c所示为,其中n可以为1、2、3或4,
[0184][0185]
在一些实施方式中,所述r
13
可以为h、c
1-6
烷基、c
1-6
取代烷基、c
3-8
环烷基或者c
3-8
取代的环烷基。
[0186]
例如,当r
13
为甲基时,r3为r1为其中n可以为1、2、3或4。
[0187]
在一些实施方式中,当r5为h,r6为时,r3可以为所述的式i的化合物可以如式i-1-1d所示为,其中n可以为1、2、3或4,
[0188][0189]
在一些实施方式中,所述r
13
可以为h、c
1-6
烷基、c
1-6
取代烷基、c
3-8
环烷基或者c
3-8
取代的环烷基。
[0190]
例如,当r
13
为甲基时,r3为r1为其中n可以为1、2、3或4。
[0191]
在一些实施方式中,r1可以选自以下取代基:
[0192][0193]
在一些实施方式中,a环可以选自以下取代芳香环:
[0194][0195]
在一些实施方式中,所述a环可以为取代的苯环、取代的吡啶环或取代的吡唑环。
[0196]
在一些实施方式中,当a环为取代的苯环或者取代的吡啶环时,r2取代位置可以位于x的对位或者间位。
[0197]
例如,a环可以选自以下结构:
[0198][0199]
在一些实施方式中,r2可以选自以下取代基:
[0200][0201]
例如,a环可以选自以下结构:
[0202][0203]
在一些实施方式中,a环可以选自以下结构:
[0204][0205]
在一些实施方式中,r
12
可以选自以下取代基:
[0206][0207]
例如,a环可以选自以下结构:
[0208][0209]
在一些实施方式中,所述式i的化合物可以选自以下化合物:
[0210]
[0211]
[0212]
[0213][0214][0215]
在一些实施方式中,所述的式i的化合物可以如式i-2所示,
[0216][0217]
或其药学上可接受的盐、溶剂化物、立体异构体、前药、代谢物或衍生物,其中,rb,rc,rd,re可以各自独立地选自h、羟基、卤素、氰基、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代的烷基、c
3-8
取代的环烷基和c
1-6
烷氧基;
[0218]
x可以选自—o—、—nh—、—s—、—so—、—so2—、羰基、羰基氨基和氨基羰基;
[0219]
r1可以为取代的环烷基、取代的杂环基或者m可以为0、1、2、3、4或5,r
14
可以为c
1-6
烷基、c
3-8
环烷基、c
1-6
取代的烷基或c
3-8
取代的环烷基;
[0220]
a环为取代的苯环或取代的5-6元芳香杂环,其中,所述取代的5-6元芳香杂环具有1-4个选自o、s和n的杂原子;
[0221]
r2可以选自以下取代基:
[0222][0223]
其中y可以为c或者n,r7可以为h、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代烷基或者c
3-8
取代的环烷基,r8可以为h、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代烷基或者c
3-8
取代的环烷基,r9和r
10
可以独立地为h、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代烷基或者c
3-8
取代的环烷基,r
11
可以为h、氨基、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代烷基或者c
3-8
取代的环烷基,r
12
可以为4-6元取代杂环基或者4-6元未取代杂环基。
[0224]
在一些实施方式中,所述的式i的化合物可以如式i-3所示,
[0225][0226]
或其药学上可接受的盐、溶剂化物、立体异构体、前药、代谢物或衍生物,其中,rb,rc,rd,re可以各自独立地选自h、羟基、卤素、氰基、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代的烷基、c
3-8
取代的环烷基和c
1-6
烷氧基;
[0227]
x可以选自—o—、—nh—、—s—、—so—、—so2—、羰基、羰基氨基和氨基羰基;
[0228]
r1可以为取代的环烷基、取代的杂环基或者m可以为0、1、2、3、4或5,r
14
可以为c
1-6
烷基、c
3-8
环烷基、c
1-6
取代的烷基或c
3-8
取代的环烷基;
[0229]
a环为取代的苯环或取代的5-6元芳香杂环,其中,所述取代的5-6元芳香杂环具有1-4个选自o、s和n的杂原子;
[0230]
r2可以选自以下取代基:
[0231][0232]
其中y可以为c或者n,r7可以为h、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代烷基或者c
3-8
取代的环烷基,r8可以为h、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代烷基或者c
3-8
取代的环烷基,r9和r
10
可以独立地为h、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代烷基或者c
3-8
取代的环烷基,r
11
可以为h、氨基、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代烷基或者c
3-8
取代的环烷基,r
12
可以为4-6元取代杂环基或者4-6元未取代杂环基。
[0233]
在一些实施方式中,所述的式i的化合物可以如式i-4所示,
[0234][0235]
或其药学上可接受的盐、溶剂化物、立体异构体、前药、代谢物或衍生物,其中,rb,rc,rd,re可以各自独立地选自h、羟基、卤素、氰基、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代的烷基、c
3-8
取代的环烷基和c
1-6
烷氧基;
[0236]
x可以选自—o—、—nh—、—s—、—so—、—so2—、羰基、羰基氨基和氨基羰基;
[0237]
r1可以为取代的环烷基、取代的杂环基或者m可以为0、1、2、3、4或5,r
14
可以为c
1-6
烷基、c
3-8
环烷基、c
1-6
取代的烷基或c
3-8
取代的环烷基;
[0238]
a环为取代的苯环或取代的5-6元芳香杂环,其中,所述取代的5-6元芳香杂环具有1-4个选自o、s和n的杂原子;
[0239]
r2可以选自以下取代基:
[0240][0241]
其中y可以为c或者n,r7可以为h、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代烷基或者c
3-8
取代的环烷基,r8可以为h、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代烷基或者c
3-8
取代的环烷基,r9和r
10
可以独立地为h、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代烷基或者c
3-8
取代的环烷基,r
11
可以为h、氨基、c
1-6
烷基、c
3-8
环烷基、c
1-6
取代烷基或者c
3-8
取代的环烷基,r
12
可以为4-6元取代杂环基或者4-6元未取代杂环基。
[0242]
药物组合物
[0243]
另一方面,本技术提供了一种药物组合物,其包含至少一种所述的式i的化合物,或其药学上可接受的盐、溶剂化物、立体异构体、前药、代谢物或衍生物;以及一种或多种无毒的药学上可接受的载体和/或稀释剂和/或辅料(在本技术中统称为“载体”材料),且当希望时,包含其它活性成分。
[0244]
不欲被任何理论所限,下文中的实施例仅仅是为了阐释本技术的化合物、制备方法和用途等,而不用于限制本技术的范围。
[0245]
实施例
[0246]
实施例1:化合物dd02001h:8-(((1s,4s)-4-氨基环己基)氧代)-n-(1-(1-甲基哌啶-4-基)-1h-吡唑-4-基)喹唑啉-2-胺的合成
[0247]
图1显示了化合物dd02001h的合成路线。
[0248]
1.1化合物2的合成
[0249]
在n2保护、0℃下,向化合物1(25.0g,150mmol,1.00eq)的thf(300ml)溶液中逐滴添加bh3/thf(1.00m,329ml,2.20eq)。滴加完毕后,将混合物在0℃下搅拌30分钟,然后将混合物加热至50℃并搅拌12小时。tlc(石油醚:乙酸乙酯=1:1,原料rf=0.1,产品rf=0.3)显示有新斑点形成,原料完全消失,反应完全,随后将混合物冷却至0℃,逐滴添加meoh(400ml)至无气泡形成,然后添加40.0ml h2o并用乙酸乙酯(300ml*2)萃取,有机层盐水(100ml*2)洗涤,无水na2so4干燥并过滤,然后减压浓缩获得化合物2(45.0g,294mmol,98.2%产率),该物质为淡黄色油状物质,结构经1h nmr证实。
[0250][0251]1h nmr(400mhz,cdcl3)δ6.65-6.85(m,3h),4.63(s,2h),3.86(s,3h)。
[0252]
1.2化合物3的合成
[0253][0254]
向化合物2(45.0g,294mmol,1.00当量)的二氯甲烷(dcm)(500ml)溶液中添加mno2(128g,1.47mol,5.00当量),所得混合物在25℃下搅拌12小时。薄层色谱(石油醚:乙酸乙酯=1:1,物料rf=0.45,产品rf=0.8)显示有新斑点形成,原料点消失,反应结束后混合物经硅藻土过滤,滤液减压浓缩。经柱层析法(sio2,石油醚/乙酸乙酯=30/1至5/1)分离纯化得化合物3(25.0g,166mmol,56.3%产率),结构经1h nmr证实。
[0255]1h nmr(400mhz,dmso-d6)δ9.85(s,1h),7.18(dd,j=8.0,1.2hz,1h),7.02(dd,j=8.0,0.8hz,1h),6.80(br s,2h),6.64(t,j=8.0hz,1h),3.77-3.87(m,3h).
[0256]
1.3化合物4的合成
[0257][0258]
将化合物3(23.0g,152mmol,1.00eq)、尿素(101g,1.67mol,89.7ml,11.0eq)和nh4oac(586mg,7.61mmol,0.05eq)的混合物在160℃下搅拌0.5小时,混合物开始从热溶液中析出沉淀。添加nmp(100ml)以溶解固体沉淀后,将反应在160℃下搅拌1小时。lc-ms显示检测到ms(rt=0.247分钟)目标产物4,原料消失,反应结束后将混合物冷却至25℃并倒入100ml h2o中,25℃下搅拌10分钟,在减压下过滤。粗产物用石油醚(60.0ml)在25℃下振荡分散5min,并在减压下过滤以获得化合物4(20.0g,114mmol,74.6%产率),为灰色固体。结构经1h nmr证实。
[0259]
lcms:产物rt=0.247min,m/z=177.2(m+h)
+
[0260]1h nmr(400mhz,dmso)δ8.36(s,1h),6.90(br d,j=4.0hz,1h),6.87(d,j=8.0hz,1h),6.79(dd,j=8.0,4.0hz,1h),5.98(dd,j=8.0,4.0hz,1h),3.79(s,3h)。
[0261]
1.4化合物5的合成
[0262]
将化合物4(19.0g,108mmol,1.00eq)于0℃下加入pocl3(248g,1.61mol,150ml,15.0eq)中,所得混合物在25℃搅拌0.5小时,然后升温至140℃搅拌2小时。lc-ms显示检测到目标产物ms(rt=0.655分钟)。反应结束后,将混合物缓慢倒入0-10℃的冰水(200ml)中并搅拌,然后用乙酸乙酯(300ml*4)萃取,盐水(100ml*2)洗涤有机层,无水na2so4干燥,过滤并减压浓缩。在25℃下用石油醚(60.0ml)振荡分散5min,过滤得黄色固体化合物5(7.60g,39.1mmol,36.2%产率),结构经1h nmr确认。
[0263][0264]
lcms:产物rt=0.655 min,m/z=195.0(m+h)
+
[0265]1h nmr(400 mhz,dmso)δ9.56(s,1h),7.69-7.79(m,2h),7.54(br d,j=4.0 hz,1h),3.99(s,3h)。
[0266]
1.5化合物6的合成
[0267][0268]
在0℃下向化合物6-1(10.0 g,86.8 mmol,10.1 ml,1.00 eq)、化合物6-1a(11.7 g,104mmol,1.20 eq)和三苯基膦烷(34.2 g,130 mmol,1.50 eq)的thf(300 ml)溶液中加入diad(26.3 g,130 mmol,25.3 ml,1.50 eq),所得混合物在25℃、n2保护下搅拌12小时。lcms显示原料反应完全,产物形成。反应结束后,用1 m hcl将反应混合物调节至ph=4,并用乙酸乙酯(300 ml)萃取,用nahco3(饱和)将水相调节至ph=8,并用乙酸乙酯(100ml*2)再次萃取。用饱和盐水(100 ml)洗涤有机层,有机层使用无水硫酸钠干燥,过滤并减压浓缩得到化合物6-2(8.00 g,38.1 mmol,43.8%产率),该化合物为黄色油状物质,结构经1h nmr证实。
[0269]
lcms产物rt=0.150 min,m/z=211.1(m+h)
+
[0270]1h nmr(400 mhz,cdcl3)δ8.15(s,1h),8.04(s,1h),4.08-4.14(m,1h),2.95-2.98(m,2h),2.31(s,3h),2.09-2.15(m,6h)。
[0271]
化合物6-2(8.00 g,38.1 mmol,1.00当量)、pd/c(2.00 g,10%纯度)在甲醇(50.0 ml)中搅拌,混合物于25℃下在h2(15 psi)下搅拌12小时。lcms显示化合物6-2被完全消耗,反应结束后将混合物过滤并减压浓缩,得到黄色油状化合物6(5.00 g,27.7 mmol,72.9%产率),结构经1h nmr证实。
[0272]
lcms:产物rt=0.10 min,m/z=181.1(m+h)
+
[0273]1h nmr(400 mhz,cdcl3)δ7.09(s,1h),7.01(s,1h),3.92-3.98(m,1h),2.88-2.91(m,4h),2.26(s,3h),1.85-2.26(m,6h)。
[0274]
1.6化合物7的合成
[0275][0276]
向化合物5(400mg,2.06mmol,1.00eq)和化合物6(444mg,2.47mmol,1.20eq)的ipa(10.0ml)中溶液中添加tfa(23.4mg,205umol,15.2ul,0.10eq),所得混合物在100℃下搅拌
2小时。lcms显示检测到目标产物质量(rt=0.653min)。用乙酸乙酯(100ml)稀释混合物并用饱和nahco3溶液(50.0ml)洗涤,再用盐水(50.0ml)洗涤有机相,无水硫酸钠干燥,过滤并浓缩得到固体物质,用石油醚(25.0ml)振荡分散得到化合物7(570mg,1.68mmol,81.9%产率),其为黄色固体,结构经lcms和1h nmr确认。
[0277]
lcms:产物rt=0.673min,m/z=339.3(m+h)
+
[0278]1h nmr(400mhz,cdcl3)δ9.05(s,1h),8.22(s,1h),7.60(s,1h),7.31-7.35(m,2h),7.21-7.25(m,1h),7.10-7.12(m,1h),4.12-4.14(m,1h),4.05(s,3h),2.98-3.01(m,2h),2.34(s,3h),2.14-2.23(m,6h).
[0279]
1.7化合物8的合成
[0280][0281]
向化合物7(570mg,1.68mmol,1.00eq)的dcm(15.0ml)溶液中逐滴添加bbr3(1.05g,4.21mmol,405.7ul,2.50eq)。在25℃下将混合物搅拌12小时。lcms(ew20001-101-p1a1)显示检测到目标产物(rt=0.622min)。反应结束周,用dcm(100ml)稀释混合物,并用饱和nahco3溶液(50.0ml)洗涤,用盐水(50.0ml)洗涤有机相,无水硫酸钠干燥,过滤并浓缩得到化合物8(500mg,粗品)作为黄色固体,在下一步中直接使用。
[0282]
lcms:产物rt=0.847min,m/z=325.2(m+h)
+
[0283]
1.8化合物10的合成
[0284][0285]
向化合物8(200mg,616umol,1.00eq)和化合物9(273mg,740umol,1.20eq)的dmf(3.00ml)溶液中添加cs2co3(502mg,1.54mmol,2.50eq),所得混合物在80℃下搅拌1小时。lcms显示检测到目标产物(rt=0.799min)。反应结束后,添加水(50.0ml)淬灭混合物并用乙酸乙酯(50.0ml*2)萃取,用盐水(50.0ml)洗涤有机相,无水硫酸钠上干燥,过滤并浓缩得到化合物10(320mg,613umol,99.5%产率),黄色油状物质,未经进一步纯化,下一步直接使用。
[0286]
lcms产物rt=0.799min,m/z=522.3(m+h)
+
。
[0287]
1.9化合物dd02001h的合成
[0288]
[0289]
在25℃下将化合物10(320mg,613umol,1.00eq)与hcl/二氧六环(4m,10.0ml,65.2eq)的混合物搅拌1小时。lcms显示检测到目标产物(rt=0.651min)。将混合物浓缩得到残留物,通过制备hplc纯化。用饱和nahco3溶液将混合物调节至ph=8,并用dcm(50.0ml*2)萃取,用盐水(50.0ml)洗涤有机相,无水硫酸钠干燥,过滤并浓缩得到化合物dd02001h,化合物结构经lcms、hplc(ew20001-106-p1a2)和1h-nmr确认,为黄色固体(27.2mg,91.7umol,14.9%产率,96.7%纯度)。
[0290]
lcms产物rt=0.644min,m/z=422.3(m+h)
+
[0291]1h nmr(400mhz,cdcl3)δ8.94(s,1h),8.14(s,1h),7.68(s,1h),7.23-7.25(m,1h),7.12-7.13(m,2h),7.06(s,1h),4.67-4.68(m,1h),4.05-4.09(m,1h),2.89-2.92(m,2h),2.78-2.80(m,1h),2.26(s,3h),2.05-2.15(m,8h),1.62-1.74(m,8h).
[0292]
实施例2:化合物dd02013h:4-((2-((4-((2-(二甲氨基)乙基)(甲基)氨基)苯基)氨基)喹唑啉-8-基)氧代)环己烷-1-醇的合成
[0293]
图2显示了化合物dd02013h的合成路线。
[0294]
2.1化合物11的合成
[0295][0296]
0℃下,向化合物5(500mg,2.57mmol,1.00eq)的dcm(10.0ml)溶液中逐滴加入bbr3(1.42g,5.65mmol,545ul,2.20eq)的dcm(5.00ml)溶液。滴加完毕,在25℃下将混合物搅拌12小时。lcms显示检测到目标产品分子量(rt=0.585min)。反应结束后,混合物加水(50.0ml)淬火并用dcm(50.0ml)萃取。用盐水(50.0ml)洗涤有机相,无水硫酸钠干燥,过滤并浓缩以得到残余物,该残留物通过柱层析法(sio2,石油醚/乙酸乙酯=20/1到10/1,tlc(石油醚:乙酸乙酯=3:1,rf=0.3)纯化,得到黄色固体化合物11(400mg,2.21mmol,86.2%产率),结构经lcms与1h nmr确认。
[0297]
lcms:产物rt=0.575min,m/z=181.0(m+h)
+
[0298]1h nmr(400mhz,cdcl3)δ9.30(s,1h),7.59-7.64(m,1h),7.49-7.52(m,1h),7.42-7.45(m,2h)。
[0299]
2.2化合物12的合成
[0300][0301]
向化合物11(200mg,1.11mmol,1.00eq)和化合物11a(639mg,1.66mmol,1.50eq)的dmf(5.00ml)溶液中添加cs2co3(721.6mg,2.21mmol,2.00eq),所得混合物在80℃下搅拌2小
时。lcms检测到目标产物分子量(rt=1.229分钟)。反应结束后,添加水(50.0ml)淬灭反应,用乙酸乙酯(50.0ml*2)萃取,盐水(50.0ml)洗涤有机相,无水硫酸钠干燥,过滤并浓缩得到残留物,通过柱层析法(sio2,石油醚/乙酸乙酯=20/1到10/1,tlc(石油醚/乙酸乙酯=5:1,rf=0.4))纯化得到黄色固体化合物12(100mg,254umol,22.9%产率),结构经1h nmr证实。
[0302]
lcms:产物rt=1.229mins,m/z=393.2(m+h)
+
[0303]1h nmr(400mhz,cdcl3)δ9.25(s,1h),7.55-7.60(m,1h),7.49-7.51(m,1h),7.34-7.37(m,1h),4.53-4.57(m,1h),3.90-3.92(m,1h),2.15-2.20(m,2h),1.86-1.89(m,4h),1.60-1.64(m,2h),0.93(s,9h),0.08(s,6h)。
[0304]
2.3化合物12a的合成
[0305][0306]
向化合物12-1(2.00g,14.2mmol,1.50ml,1.00eq)和化合物12-1a(1.59g,15.6mmol,2.03ml,1.10eq)的二甲基亚砜(10.0ml)溶液中添加k2co3(3.92g,28.4mmol,2.00eq),所得混合物在40℃下搅拌2小时。tlc(石油醚:乙酸乙酯=5:1)显示化合物12-1(rf=0.6)保留,并检测到新产物形成。反应结束后,用h2o(100ml)稀释反应混合物并用乙酸乙酯(100ml*2)萃取,饱和盐水(200ml)洗涤合并的有机层,无水硫酸钠干燥,过滤并减压浓缩,得到化合物12-2(3.20g,原油),结构经1h nmr确认后用于下一步骤,未进行进一步的纯化。
[0307]1h nmr(400mhz,cdcl3)δ8.09-8.12(m,2h),6.59-6.62(m,2h),3.55(t,j=7.2hz,2h),3.10(s,1h),2.50(t,j=7.2hz,2h),2.30(s,6h)。
[0308]
向化合物12-2(3.20g,14.3mmol,1.00当量)的meoh(30.0ml)溶液中添加pd/c(1.00g,10%纯度,1.00当量)。悬浮液在真空下脱气,并用氢气吹扫几次,之后所得溶液在25℃的h2(15psi)氛围下搅拌2小时。tlc(二氯甲烷:甲醇=10:1)显示化合物12-2(rf=0.5)原料点消失,并检测到新化合物形成。反应结束后,将混合物过滤并浓缩以得到化合物12a(2.30g,11.9mmol,83.0%产率),产物为红棕色油,结构经1h nmr确证,未经进一步纯化。
[0309]1h nmr(400mhz,dmso)δ6.47-6.54(m,4h),4.34(br.s,2h),3.19(t,j=7.2hz,2h),2.72(s,1h),2.30(t,j=7.2hz,2h),2.14(s,6h)。
[0310]
2.4化合物13的合成
[0311]
[0312]
向化合物12(100mg,254umol,1.00eq)和化合物12a(59.0mg,305umol,1.20eq)的ipa(3.00ml)溶液中添加tfa(29.0mg,255umol,18.8ul,1.00eq)。所得混合物在100℃下搅拌1小时。lcms显示检测到目标产物分子量(rt=0.913min)。反应结束后,用乙酸乙酯(100ml)萃取,先后用饱和nahco3溶液(50.0ml)、盐水(50.0ml)洗涤有机相,无水硫酸钠干燥,过滤并浓缩,得到化合物13(130mg,粗品),黄色固体,未纯化,直接用于下一步。
[0313]
2.5化合物dd02013h的合成
[0314][0315]
向化合物13(130mg,236umol,1.00当量)的dcm(5.00ml)溶液中添加tfa(0.50ml),所得混合物在25℃下搅拌1小时。lcms显示检测到目标产物分子量(rt=0.701min)。反应结束后,将混合物浓缩得到残留固体,通过制备高效液相色谱法纯化,所得固体用饱和nahco3溶液调节至ph=8,dcm(50.0ml*2)萃取,用盐水(50.0ml)洗涤,无水硫酸钠干燥,过滤并浓缩,得黄色固体、目标化合物dd02013h(47.6mg,130.7umol,55.3%产率,94.9%纯度),结构经lcms、hplc和1h-nmr确认。
[0316]
lcms产物rt=0.997min,m/z=436.3(m+h)
+
[0317]
hplc产物rt=2.951mins,purity:95.6%
[0318]1h nmr(400mhz,cdcl3)δ9.00(s,1h),7.83-7.85(m,2h),7.29-7.31(m,1h),7.17-7.19(m,3h),6.85-6.87(m,2h),4.78-4.79(m,1h),3.76-3.81(m,1h),3.49(t,j=7.2hz,2h),2.95(s,3h),2.54(t,j=7.2hz,2h),2.33(s,6h),2.16-2.21(m,2h),2.03-2.06(m,2h),1.80-1.83(m,2h),1.69-1.73(m,2h).
[0319]
实施例3:化合物dd02014h:4-((2-((1-(1-甲基哌啶-4-基)-1h-吡唑-4-基)氨基)喹唑啉-8-基)氧代)环己烷-1-醇的合成
[0320]
图3显示了化合物dd02014h的合成路线。
[0321]
向化合物8(400mg,1.23mmol,1.00eq)和化合物11a(569.1mg,1.48mmol,1.20eq)的dmf(10.0ml)溶液中添加cs2co3(1.00g,3.08mmol,2.50eq),所得混合物在80℃下搅拌1小时。lcms显示检测到目标产物分子量(rt=0.958min)。反应结束后,加入乙酸乙酯(100ml),并先后用饱和nahco3溶液(50.0ml)、盐水(50.0ml)洗涤有机相,无水硫酸钠干燥,过滤并浓缩得到残余物,所述残留物用石油醚:乙酸乙酯(3:1,20.0ml)振荡分散,过滤并干燥,得到黄色固体、化合物14(300mg,559umol,45.3%产率),结构经lcms表征。
[0322]
lcms产物rt=0.912min,m/z=537.3(m+h)
+
。
[0323]
在25℃下将化合物14(300mg,559umol,1.00eq)与hcl/meoh(4m,10.0ml,71.6eq)的混合物搅拌1小时,lcms(ew20001-117-p1a1)显示检测到产物消失、目标产物分子量出现(rt=0.729min)。反应结束后,将混合物浓缩,所得固体经制备hplc纯化,随后用饱和nahco3溶液将混合物调节至ph=8,dcm(50.0ml*2)萃取,盐水(50.0ml)洗涤有机相,无水硫酸钠干燥,过滤并浓缩得黄色固体、化合物dd02014h(35.5mg,138umol,24.6%产率,96.9%
纯度),结构经lcms、hplc和1h nmr确证。
[0324]
lcms产物rt=0.730min,m/z=423.2(m+h)
+
[0325]
hplc产物rt=1.192mins,purity:98.0%
[0326]1h nmr(400mhz,cdcl3)δ9.01(s,1h),8.61(s,1h),7.49(s,1h),7.28-7.30(m,1h),7.17-7.22(m,3h),4.66-4.68(m,1h),4.23-4.30(m,1h),3.85-3.89(m,1h),2.99-3.03(m,2h),2.33(s,3h),2.25-2.29(m,4h),2.15-2.17(m,4h),1.95-2.06(m,2h),1.84-1.88(m,4h).
[0327]
实施例4:化合物dd02006h:4-((2-((4-(4-甲基哌嗪-1-基)苯基)氨基)喹唑啉-8-基)氧代)环己烷-1-醇的合成
[0328]
图4显示了化合物dd02006h的合成路线。
[0329]
4.1化合物16的合成
[0330]
25℃下,向化合物5(1.00g,5.14mmol,1.00eq)和化合物16a(1.08g,5.65mmol,1.10eq)的ipa(20.0ml)溶液中添加tfa(586mg,5.14mmol,380ul,1.00eq),混合均匀后,将反应混合物加热至100℃并搅拌反应3小时。
[0331][0332]
lcms显示检测到目标产物的分子量(rt=0.691分钟)。反应结束后,将混合物倒入30.0ml h2o中,然后用乙酸乙酯(60.0ml*4)萃取,盐水(60.0ml*2)洗涤有机层,无水na2so4干燥,过滤并减压浓缩,得到化合物16(1.50g,粗品),为一黄色固体。产品未经纯化,直接用作下一步。
[0333]
lcms:产物rt=0.691min,m/z=350.2(m+h)
+
[0334]
4.2化合物17的合成
[0335][0336]
0℃下,向化合物16(1.30g,3.72mmol,1.00eq)的dcm(20.0ml)溶液中逐滴添加溶解于dcm(10.0ml)中的bbr3(2.80g,11.2mmol,1.08ml,3.00eq),滴加完毕之后,在0℃搅拌30min,随后将混合物加热至25℃并搅拌12h。lcms显示检测到目标产物的分子量(rt=0.677min)。反应结束后,将混合物倒入冰水(30.0ml)中,用乙酸乙酯(60.0ml*3)萃取,将盐水洗涤有机层(30.0ml*2),无水na2so4上干燥,过滤并减压浓缩,得到化合物17(1.20g,粗
品),为一灰色固体。产品未经纯化,直接用作下一步。
[0337]
lcms:产物rt=0.677min,m/z=336.2(m+h)
+
[0338]
4.3化合物18的合成
[0339][0340]
化合物17(150mg,447umol,1.00eq)、化合物11a(276mg,894umol,2.00eq)和cs2co3(364mg,1.12mmol,2.50eq)溶解于dmf(10.0ml)中,随后将混合物加热到80℃反应12小时。lcms显示检测到目标产品分子量(rt=0.934分钟)。反应结束后,将混合物倒入30.0ml h2o中,然后用乙酸乙酯(30.0ml*4)萃取,将有机层盐水(30.0ml*2)洗涤,无水na2so4干燥,过滤并减压浓缩,得到化合物18(200mg,粗品),为一黄色固体。产品未经纯化,直接用作下一步。
[0341]
lcms产物:rt=0.934min,m/z=548.4(m+h)
+
[0342]
4.4化合物dd02006h的合成
[0343][0344]
在25℃下向化合物18(200mg,365umol,1.00eq)的dcm(6.00ml)溶液中滴加hcl/1,4-二氧六环(4m,2.00ml,21.9eq),所得混合物搅拌2小时。lcms显示检测到目标产物分子量(rt=0.746min)。薄层色谱(dcm:meoh=8:1,物料rf=0.5,产品rf=0.2)显示有新斑点形成,无残留物质。将混合物倒入30.0ml h2o中,用饱和nahco3调节ph值约8,然后用乙酸乙酯(30.0ml*4)萃取,盐水(30.0ml*2)洗涤有机层,无水na2so4干燥,过滤并减压浓缩。粗产物经预高效液相色谱,减压浓缩,得到黄色固体目标产物(68.6mg,157umol,43.1%产率,99.4%纯度),结构经1h nmr、lcms和hplc确认。
[0345]
lcms产物:rt=0.734min,m/z=434.3(m+h)
+
;
[0346]
hplc产物rt=1.373mins,99.4%纯度;
[0347]1h nmr(400mhz,dmso)δ9.57-9.71(m,1h),9.12-9.27(m,1h),7.90-8.06(m,2h),7.40-7.47(m,1h),7.28-7.34(m,1h),7.18-7.26(m,1h),6.86-6.99(m,2h),4.75(br s,1h),4.52-4.64(m,1h),3.63(br s,1h),3.07(br d,j=4.0hz,4h),2.24(s,3h),1.91-2.05(m,2h),1.74-1.87(m,2h),1.56-1.72(m,4h).
[0348]
实施例5:化合物dd02008h:8-(2-甲氧基乙氧基)-n-(4-(4-甲基哌嗪-1-基)苯基)喹唑啉-2-胺的合成
[0349]
图5显示了化合物dd02008h的合成路线。
[0350]
将化合物17(150mg,447umol,1.00eq)、化合物17a(68.4mg,492umol,46.2ul,1.10eq)和cs2co3(437mg,1.34mmol,3.00eq)溶解于dmf(10.0ml)中,然后将混合物加热至80℃并搅拌反应12小时,lcms显示检测到目标产品分子量(rt=0.703分钟)。薄层色谱(dcm:meoh=10:1,物料rf=0.1,产品rf=0.3)显示有新斑点形成,无起始原料。反应结束后,将混合物倒入30.0ml h2o中,然后用乙酸乙酯(30.0ml*4)萃取,盐水(30.0ml*2)洗涤有机层,无水na2so4干燥,过滤并减压浓缩。通过柱层析法(sio2,dcm/meoh=20/1至5/1)纯化,获得黄色固体目标化合物dd02008h(71.94mg,183umol,40.9%产率,98.6%纯度),结构经1h nmr、lcms和hplc确证。
[0351]
lcms产物:rt=0.710min,m/z=416.2(m+h+na)
+
[0352]
hplc产物:rt=1.267min,98.7%purity
[0353]1h nmr(400mhz,cdcl3)δ9.04(s,1h),7.72(br d,j=8.0hz,2h),7.33(dd,j=8.0,4.0hz,1h),7.15-7.24(m,2h),6.97(d,j=8.0hz,2h),4.33-4.41(m,2h),3.91-3.99(m,2h),3.56(s,3h),3.13-3.25(m,4h),2.58-2.65(m,4h),2.38(s,3h).
[0354]
实施例6:化合物dd02015h:3-((2-((4-(4-甲基哌嗪-1-基)苯基)氨基)喹唑啉-8-基)氧代)环戊烷-1-醇的合成
[0355]
图6显示了化合物dd02015h的合成路线。
[0356]
将化合物17(150mg,447umol,1.00eq)、化合物18a(331mg,894umol,2.00eq)和cs2co3(364mg,1.12mmol,2.50eq)溶解于dmf(10.0ml)中,然后将混合物加热至80℃并在搅拌反应12小时。lcms显示检测到目标产品分子量(rt=0.893min),tlc(dcm:meoh=10:1,物料rf=0.1,产品rf=0.25)显示有新斑点形成,无起始原料。反应结束后,所得混合液倒入30ml水中,随后用乙酸乙酯(30.0ml*4)萃取,盐水洗涤(30.0ml*2),无水硫酸钠干燥,过滤,减压除去溶剂,所得残留固体通过柱层析法(sio2,dcm/meoh=20/1至5/1)纯化,得到黄色固体化合物19(200mg,375umol,83.8%产率)。lcms产物:rt=0.893 min,m/z=534.4(m+h)
+
[0357]
将化合物19(200 mg,375 umol,1.00 eq)溶于dcm(6.00 ml)中,25℃下滴加hcl/1,4-二氧六环(4 m,4.00 ml,42.7 eq),滴加完毕,搅拌反应2小时。lcms显示检测到目标产物分子量(rt=0.721min),高效液相色谱显示起始原料反应完毕。反应结束后将混合物倒入30.0 ml h2o中,用饱和nahco3调节至ph值约8,然后用乙酸乙酯(30.0 ml*4)萃取,有机层盐水(30.0 ml*2)洗涤,无水na2so4干燥,过滤并减压浓缩,所得粗产物经制备高效液相色谱纯化,得黄色固体目标化合物dd02015h(110 mg,262 umol,70.0%产率,100%纯度),结构经1h nmr、、lcms和hplc确证。
[0358]
lcms产物:rt=0.716 min,m/z=420.3(m+h)
+
[0359]
hplc产物:rt=1.286 min,100%纯度.
[0360]1h nmr(400 mhz,cdcl3)δ9.04(s,1 h),7.60(br d,j=8.0 hz,2 h),7.32-7.38(m,1 h),7.21-7.24(m,2 h),6.93-7.01(m,2 h),5.15(t,j=4.0 hz,1 h),4.41(br t,j=4.0 hz,1 h),3.17-3.26(m,4 h),2.62(m,4 h),2.38(s,3 h),1.94-2.20(m,6 h).
[0361]
实施例7:化合物dd02021h:4-((2-((5-(4-甲基哌嗪-1-基)吡啶-2-基)氨基)喹唑啉-8-基)氧代)环己烷-1-醇的合成
[0362]
图7显示了化合物dd02021h的合成路线。
[0363]
7.1化合物20的合成
[0364]
在25℃下,将化合物5(500 mg,2.57 mmol,1.00 eq)、化合物19a(988 mg,5.14 mmol,2.00 eq)、pd2(dba)3(353 mg,386 umol,0.15 eq)和binap(184 mg,295 umol,1.15e-1 eq)溶解于1,4-二氧六环(30.0 ml)中,所得混合物中加入cs2co3(1.67 g,5.14 mmol,2.00 eq),然后将反应混合物加热到100℃
[0365][0366]
并搅拌反应10小时。lcms显示检测到目标产物分子量(rt=0.643 min)。反应结束后,将混合物倒入30.0 ml h2o中,然后用dcm(50.0 ml*3)萃取,有机层盐水(30.0 ml*3)洗涤,无水na2so4上干燥,过滤并减压浓缩,得到化合物20(600 mg,粗品),为一黄色固体。产品未经纯化,直接用于下一步反应。
[0367]
7.2化合物21的合成
[0368][0369]
将化合物20(600mg,1.71mmol,1.00eq)溶于dcm(10.0ml)中,所得溶液逐滴添加溶解于dcm(5.00ml)中的bbr3(429mg,1.71mmol,165ul,1.00eq),滴加完毕,体系在25℃下搅拌反应12小时。lcms显示检测到目标产物分子量(rt=0.321min)。反应结束后,将混合物倒入冰水(40.0ml)中,用饱和nahco3将ph调整至约8,然后用dcm(50.0ml*3)萃取,有机层经盐水(30.0ml*3)洗涤,无水硫酸钠干燥,过滤并减压浓缩,得到化合物21(600mg,粗品),为一黄色固体。产品未经纯化,直接用于下一步反应。lcms产物rt=0.324min,m/z=337.2(m+h)
+
[0370]
7.3化合物22的合成
[0371][0372]
将化合物21(600mg,1.78mmol,1.00eq)、化合物11a(1.37g,3.57mmol,2.00eq)和cs2co3(1.45g,4.46mmol,2.50eq)溶解于dmf(10.0ml)中,所得混合物加热到80℃反应12小时。lcms显示检测到目标产物分子量(rt=0.830min),薄层色谱(dcm:meoh=10:1,物料rf=0.1,产品rf=0.3)显示有新斑点形成,无起始原料。反应结束后,将混合物倒入30.0ml h2o中,然后用乙酸乙酯(60.0ml*3)萃取,有机层盐水(40.0ml*2)洗涤,无水na2so4干燥,过滤并减压浓缩,所得物质通过柱层析法(sio2,dcm/meoh=40/1至5/1)纯化,得到化合物22
(700mg,1.08mmol,60.5%产率,84.6%纯度),为一黄色固体。
[0373]
lcms产物rt=0.830min,m/z=549.4(m+h)
+
[0374]
hplc产物rt=2.563mins,84.6%纯度.
[0375]
7.4化合物dd02021h的合成
[0376][0377]
将化合物22(600mg,925umol,1.00当量)、hcl/1,4-二氧六环(4m,6.00ml,25.9当量)溶解于dcm(10.0ml)中,所得混合物在25℃下搅拌反应1小时。lcms显示检测到目标产物分子量(rt=0.680min),薄层色谱(dcm:meoh=10:1,物料rf=0.3,产品rf=0.15)显示有新斑点形成,无起始原料。反应结束后,搅拌下将混合物缓慢倒入40.0ml h2o中,然后用乙酸乙酯(80.0ml*3)萃取,有机层盐水(50.0ml*2)洗涤,无水na2so4干燥,过滤并减压浓缩。所得固体经制备高效液相色谱纯化,所得物质用饱和nahco3调节ph至8左右,然后用dcm(40.0ml*4)萃取,盐水(40.0ml*3)洗涤有机层,无水na2so4上干燥,减压过滤浓缩,得到目标产物dd02021h,(148mg,329umol,35.6%产率,96.7%纯度),为黄色固体,结构经1h nmr、lcms和hplc确证。
[0378]
lcms产物rt=0.717min,m/z=435.3(m+h)
+
[0379]
hplc产物rt=1.232min,96.7%纯度
[0380]1h nmr(400mhz,dmso)δ9.74(s,1h),9.26(s,1h),8.86(d,j=8.0hz,1h),8.02(d,j=4.0hz,1h),7.43-7.57(m,2h),7.24-7.40(m,2h),4.78(br s,1h),4.62-4.73(m,1h),3.54-3.69(m,1h),3.08-3.16(m,4h),2.47(m,3h),2.23(s,3h),1.93-2.04(m,2h),1.74-1.88(m,2h),1.58-1.72(m,4h).
[0381]
实施例8:化合物dd02018h:4-((8-((4-羟基环己基)氧代)喹唑啉-2-基)氨基)-n-(1-甲基哌啶-4-基)苯酰胺的合成
[0382]
图8显示了化合物dd02018h的合成路线。
[0383]
25℃下,将化合物11(400mg,2.21mmol,1.00eq)、化合物11a(1.28g,3.32mmol,1.50eq)和cs2co3(1.44g,4.43mmol,2.00eq)溶解于在dmf(10.0ml)中,所得混合物加热至80℃并搅拌反应2小时。lcms显示检测到目标产物分子量(rt=1.218min)。反应结束后,将混合物缓慢倒入40.0ml h2o中,然后用dcm(80.0ml*3)萃取,有机层盐水(50.0ml*2)洗涤,无水na2so4上干燥,过滤并减压浓缩,得到化合物15(310mg,粗品),为一灰色固体。产品未经纯化,直接用于下一步反应。
[0384]
将化合物15(260mg,661umol,1.00eq)、化合物15a(231.53mg,992.38umol,1.50eq)和tfa(75.4mg,661umol,49.0ul,1.00eq)溶解于ipa(10.0ml)中,然后将所得混合物加热至80℃并搅拌反应2小时,再将混合物加热至100℃并搅拌2小时。lcms显示检测到目标产物分子量(rt=0.762min),高效液相色谱显示纯度为59.7%(rt=1.535min)。反应结束后,在搅拌下将混合物缓慢倒入40.0ml h2o中,用饱和nahco3调节ph至8左右,然后用dcm
(80.0ml*3)萃取,有机层用盐水(50.0ml*2)洗涤,无水硫酸钠干燥,过滤并减压浓缩,所得粗品经制备高效液相色谱纯化,所得物质用饱和nahco3调节ph至8左右,用dcm(40.0ml*4)萃取,用盐水(40.0ml*3)洗涤有机层,无水na2so4干燥,过滤,浓缩,获得目标产物dd02018h(17.1mg,35.13umol,5.31%产率,97.7%纯度),为一浅黄色固体,结构经1h nmr、lcms和hplc确证。
[0385]
lcms产物rt=0.771min,m/z=476.2(m+h)
+
[0386]
hplc产物rt=1.535mins,97.3%纯度
[0387]1h nmr(400mhz,dmso-d6)δ10.15(br s,1h),9.30(br s,1h),8.23(br d,j=8.0hz,2h),8.13(br s,1h),7.83(br d,j=8.0hz,2h),7.49(br s,1h),7.27-7.42(m,2h),4.73(br s,1h),4.60(br s,1h),3.87(br s,1h),3.07(br s,2h),2.62-2.67(m,2h),2.33(br s,3h),1.84-2.08(m,3h),1.73(m,9h).
[0388]
实施例9:化合物dd02002h:4-((8-((3-羟基环戊基)氧代)喹唑啉-2-基)氨基)苯磺酰胺的合成
[0389]
图9显示了化合物dd02002h的合成路线。
[0390]
9.1化合物23的合成
[0391]
氮气保护下,将化合物11(200mg,1.11mmol)溶于无水四氢呋喃(5ml)中,依次加入三苯基磷(581mg,2.22mmol),diad(449mg,2.22mmol),搅拌15min,最后加入1,3-环戊二醇(即化合物22,340.1mg,3.33mmol),反应液室温搅拌过夜,tlc(pe/ea=3:1)分析显示原料消耗完毕,有新产物点生成。反应结束后,将所得混合物浓缩,除去溶剂,粗产品经硅胶柱分离得化合物23(210mg),收率71.5%,为一浅黄色固体。
[0392]
lcms产物rt=2.76min,m/z=265.1(m+h)
+
[0393]
9.2化合物dd02002h的合成
[0394]
氮气保护下,将化合物23(200mg,0.76mmol)溶于异丙醇(5ml)中,加入对氨基苯磺酰胺(260.2mg,1.51mmol),反应加热至90℃反应过夜,薄层层析分析显示有部分原料剩余,有新产物点生成,lcms显示检测到目标产物分子量。反应结束后,浓缩,所得粗品经硅胶柱分离,得目标分子dd02002h(104mg),收率34%,为一白色固体,结构经1h nmr和lcms确证。
[0395]
lcms产物rt=3.46min,m/z=401.1(m+h)
+
[0396]1h nmr(400mhz,meod)δ9.21(s,1h),8.23-8.25(m,2h),7.87-7.89(m,2h),7.48(d,j=7.6hz,1h),7.33-7.37(m,2h),4.36(m,1h),4.20(m,1h),1.76-2.10(m,6h)。
[0397]
实施例10:化合物dd02019h:3-((8-((4-羟基环己基)氧代)喹唑啉-2-基)氨基)苯磺酰胺的合成
[0398]
图10显示了化合物dd02019h的合成路线。
[0399]
10.1化合物26的合成
[0400]
氮气保护下,将化合物11(200mg,1.11mmol)溶于无水四氢呋喃(5ml)中,依次加入三苯基磷(581mg,2.22mmol),diad(449mg,2.22mmol),所得混合物搅拌15min,最后加入1,4-环己二醇(387mg,3.33mmol),所得反应液室温搅拌过夜,tlc(pe/ea=3:1)分析显示原料消耗完毕,有新产物点生成,lcms检测到目标产品分子量(rt=4.03min),反应液浓缩,所得粗产品经硅胶柱分离得化合物26(197mg),收率70.7%,浅黄色固体。lcms产物rt=4.03min,m/z=279.1(m+h)
+
[0401]
10.2化合物dd02019h的合成
[0402]
氮气保护下,将化合物26(150mg,0.54mmol)溶于异丙醇(5ml)中,加入间氨基苯磺酰胺(185.4mg,1.1mmol),反应加热至90℃反应过夜,tlc分析显示原料有剩余,同时有新产物点生成,lc-ms检测到目标产物分子量,反应液浓缩,所得粗品经硅胶柱分离,得目标分子dd02019h(27mg),收率12.1%,为一白色固体,结构经lc-ms与1h nmr确证。lcms产物rt=3.54min,m/z=415.2(m+h)
+
[0403]1h nmr(400mhz,meod)δ9.19(s,1h),8.51(m,2h),7.34-7.55(m,5h),4.62(m,1h),3.73(m,1h),1.57-1.94(m,8h)。
[0404]
实施例11:化合物体外抑制hpk1激酶活性的测试
[0405]
表1列出了实验所需试剂及其来源情况
[0406][0407][0408]
所有化合物测试的起始浓度为10μm,三倍浓度梯度稀释,共10个浓度点,每个浓度重复一次。
[0409]
11.1化合物体外抑制hpk1激酶活性测试
[0410]
实验开始前,配置好50μm dtt以及酶促反应系统缓冲液。
[0411]
移液枪将化合物稀释液转移到384孔板中,加毕,将384孔板密封,转速1000g下离心1min;在激酶缓冲液中,配置好2倍浓度的hpk1溶液,随后向384孔加入2.5μl配置好的2倍浓度的hpk1溶液,在转速1000g下离心30s,室温孵育10min;在激酶缓冲液中,制备2倍浓度的mbp和atp的混合液,向上述反应体系中加入2.5μl配置好的2倍浓度的mbp和atp的混合液,反应开始,在转速1000g下离心30s,随后室温孵育1h;向反应体系中加入5μl adp-glo试剂,室温孵育40min;加入10μl激酶检测试剂,室温孵育40min,随后在envision 2104plate阅读器上读取发光信号,按照以下公式计算抑制率:
[0412]
%inhibition=100-(signal
cmpd-signal
ave_pc
)/(signal
ave_vc-signal
ave_pc
)
×
100
[0413]
上式中,cmpd指测试化合物,pc指阳性对照,vc指阴性对照。hpk1实验所采用的阳性对照为sunitinib。
[0414]
表2列出了各实施例所述的化合物对hpk1激酶活性的抑制能力
[0415][0416]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1